2 releases
| 0.1.5 | Dec 17, 2024 |
|---|---|
| 0.1.4 | Mar 12, 2024 |
#41 in Biology
107 downloads per month
680KB
2K
SLoC
SeqSizzle is a pager for viewing FASTQ files with fuzzy matching, allowing different adaptors to be colored differently.
Installation
Pre-built binary
![]()
You can simply download and run the binary from Github Actions.
Conda
SeqSizzle is also available on bioconda:
conda install -c bioconda -c conda-forge seqsizzle
Cargo (crates.io)
If you already have a Rust environment set up, you can use the cargo install command:
cargo install seqsizzle
Cargo will build the seqsizzle binary and place it in $HOME/.local/share/cargo/bin/seqsizzle.
Cargo (git)
If you already have a Rust environment set up, you can use the cargo install command in your local clone of the repo:
git clone https://github.com/ChangqingW/SeqSizzle
cd SeqSizzle
cargo install --path .
Cargo will build the seqsizzle binary and place it in $HOME/.cargo.
Usage
./seqsizzle -h:
Usage: seqsizzle [OPTIONS] <FILE> [COMMAND]
Commands:
summarize Summarize the reads with patterns specified by the --patterns argument or the adapter flags. Make sure you supply the flags BEFORE the subcommand, e.g. `./SeqSizzle my.fastq -p my_patterns.csv --adapter-3p summarize`. '..' indicats unmatched regions of positive length, '-' indicates the patterns are overlapped, print the number of reads that match each pattern combination in TSV format. To be moved to the UI in the future
help Print this message or the help of the given subcommand(s)
Arguments:
<FILE> The FASTQ file to view
Options:
--adapter-3p
Start with 10x 3' kit adaptors:
- Patrial Read1: CTACACGACGCTCTTCCGATCT (and reverse complement)
- Partial TSO: AGATCGGAAGAGCGTCGTGTAG (and reverse complement)
- Poly(>10)A/T
--adapter-5p
Start with 10x 5' kit adaptors
- Patrial Read1: CTACACGACGCTCTTCCGATCT (and reverse complement)
- TSO: TTTCTTATATGGG (and reverse complement)
- Patrial Read2: AGATCGGAAGAGCACACGTCTGAA (and reverse complement)
- Poly(>10)A/T
-p, --patterns <PATTERNS_PATH>
Start with patterns from a CSV file
Must have the following header:
pattern,color,editdistance,comment
-s, --save-patterns <SAVE_PATTERNS_PATH>
Save the search panel to a CSV file before quitting. To be removed in the future since you can now hit Ctrl-S in the search panel to save the patterns
-h, --help
Print help
-V, --version
Print version
Navigation
Viewer mode
 Up / down arrow (or
Up / down arrow (or j / k) to scroll by one line, Ctrl+U / Ctrl+D to scoll half a screen.
/ (or Ctrl+F) to toggle search panel, q to quit
search panel mode
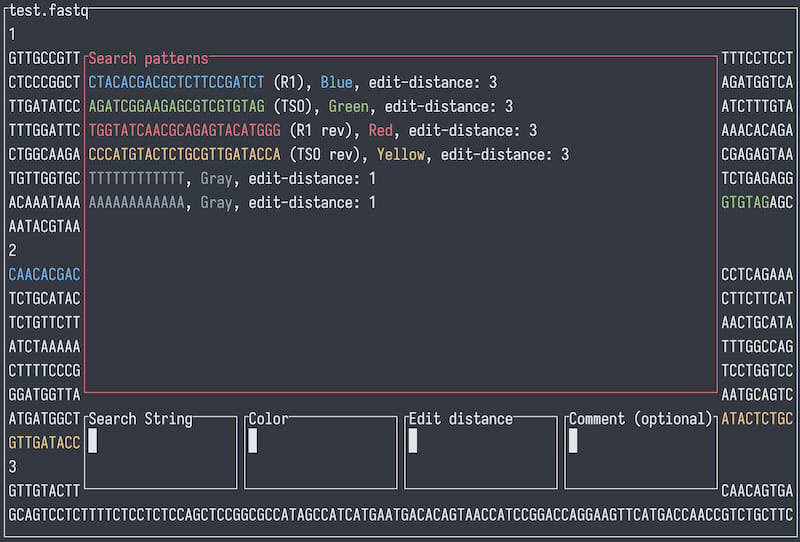 Left / right arrow (or Tab / Shift-Tab) to cycle through different input fields and the patterns list.
Left / right arrow (or Tab / Shift-Tab) to cycle through different input fields and the patterns list.
When on the patterns list field, up / down arrows cycle through patterns, Backspace (or Delete, d) to delete the selected pattern and Return to pop the pattern into the input fields for editing.
Return to add current inputs into the search pattern list (when focusing on any of the input boxes, rather than the patterns list).
Use Shift + arrow keys to move cursor within an input field (as arrow keys alone are bind to cycling input fields).
/ or Esc to close the search panel.
Roadmap
functionality
- Gzip (
fastq.gz) support - Filter reads by match
- Counting reads with match
UI
- Make elements in the search panel clickable, try implementations discussed in ratatui repo
Misc
- Unit tests
Dependencies
~36–48MB
~859K SLoC