14 releases
| 0.4.0-alpha.0 | Oct 19, 2020 |
|---|---|
| 0.3.4 | Mar 14, 2025 |
| 0.3.3 | Jan 28, 2025 |
| 0.3.2 | Aug 7, 2023 |
| 0.1.0 | Jul 31, 2017 |
#4 in Biology
1,769 downloads per month
Used in 41 crates
(37 directly)
255KB
5K
SLoC
FASTA and FASTQ parsing and writing in Rust.
![]()

Note: the master branch contains the development for version 0.4.0. The currently stable version is on a separate branch.
This library provides readers for the the following sequence formats:
- FASTA
- FASTQ (including multi-line FASTQ)
- "FASTX": Automatic recognition of the sequence format (either FASTA or FASTQ)
View documentation (for 0.4.0-alpha.x version)
Features
- Fast readers that minimize the use of allocations and copying of memory
- Flexible methods for writing FASTA and FASTQ
- Informative errors with exact positional information
- Support for recording the position and seeking back
- Serde support (for owned data structures)
- Functions for parallel processing
- Thoroughly tested using fuzzing techniques see here
Simple example
Reads FASTA sequences from STDIN and writes them to STDOUT if their length is > 100. Otherwise it prints a message.
use seq_io::fasta::{Reader,Record};
use std::io;
let mut reader = Reader::new(io::stdin());
let mut stdout = io::stdout();
while let Some(result) = reader.next() {
let record = result.expect("reading error");
// determine sequence length
let seqlen = record.seq_lines()
.fold(0, |l, seq| l + seq.len());
if seqlen > 100 {
record.write_wrap(&mut stdout, 80).expect("writing error");
} else {
eprintln!("{} is only {} long", record.id().expect("not UTF-8"), seqlen);
}
}
Records are directly borrowing data from the internal buffered reader,
no further allocation or copying takes place.
As a consequence, the while let construct has to be used instead of a for
loop.
seq_lines() directly iterates over the sequence lines, whose position
is remembered by the record, again without further copying.
Note: Make sure to add lto = true to the release profile in Cargo.toml
because calls to functions of the underlying buffered reader
(buf_redux) are not inlined otherwise.
Similar projects in Rust
- Rust-Bio: Binformatics library that provides simple and easy to use FASTA and FASTQ readers.
- fastq-rs: Very fast FASTQ parser.
seq_iowas inspired byfastq_rsin many ways. - Needletail: FASTA, FASTQ, FASTX
- fasten implements its own FASTQ reader
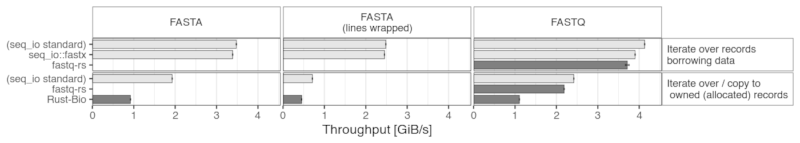
Performance comparisons
The following bar chart shows the results of a few benchmarks on random sequences generated in memory (FASTA sequences either on a single line or wrapped to a width of 80).
The readers from this crate are also compared with fastq-rs and Rust-Bio parsers. The latter is only present in the "owned" section, since there is no possibility to iterate without allocating records.
More benchmarks can be found on a separate page.
Dependencies
~0.9–1.8MB
~33K SLoC