5 releases
| new 0.1.5 | May 6, 2025 |
|---|---|
| 0.1.4 | Apr 14, 2025 |
| 0.1.3 | Apr 14, 2025 |
| 0.1.2 | Feb 13, 2025 |
| 0.1.1 | Feb 3, 2025 |
#130 in Biology
450 downloads per month
Used in 4 crates
75KB
1.5K
SLoC
paraseq
A high-performance Rust library for parallel processing of FASTA/FASTQ sequence files, optimized for modern hardware and large datasets.
Summary
paraseq is built specifically for processing paired-end FASTA/FASTQ sequences in a parallel manner.
It uses RecordSets as the primary unit of buffering, with each set directly reading a fixed number of records from the input stream.
To handle incomplete records it uses the shared Reader as an overflow buffer.
It adaptively manages buffer sizes based on observed record sizes, estimating the required space for the number of records in the set and minimizing the number of copies between buffers.
paraseq is most efficient in cases where the variance in record sizes is low as it can accurately minimize the number of copies between buffers.
It matches performance of non-record-set parsers (like seq_io and needletail), but can get higher-throughput in record-set contexts (i.e. multi-threaded contexts) by skipping a full copy of the input.
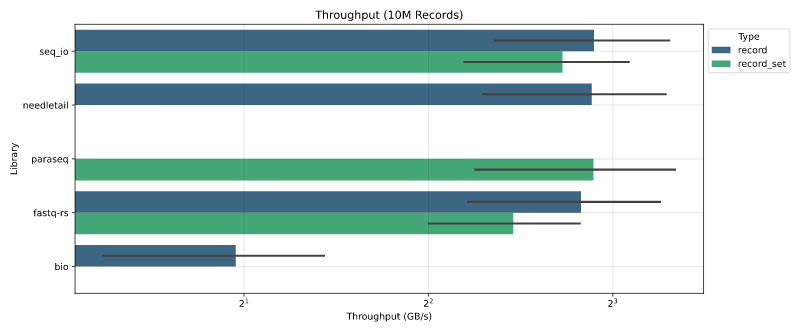 See benchmarking repo for benchmarking implementation.
See benchmarking repo for benchmarking implementation.
If you're interested in reading more about it, I wrote a small blog post discussing its design and motivation.
Usage
The benefit of using paraseq is that it makes it easy to distribute paired-end records to per-record and per-batch processing functions.
Code Examples
Check out the examples directory for code examples or the API documentation for more information.
Feel free to also explore seqpls a parallel paired-end FASTA/FASTQ sequence grepper to see how paraseq can be used in a non-toy example.
Basic Usage
This is a simple example using paraseq in a single-threaded context.
use std::fs::File;
use paraseq::fastq::{Reader, RecordSet};
use paraseq::fastx::Record;
fn main() -> Result<(), paraseq::fastq::Error> {
let file = File::open("./data/sample.fastq")?;
let mut reader = Reader::new(file);
let mut record_set = RecordSet::new(1024); // Buffer up to 1024 records
while record_set.fill(&mut reader)? {
for record in record_set.iter() {
let record = record?;
// Process record...
println!("ID: {}", record.id_str());
}
}
Ok(())
}
Parallel Processing
To distribute processing across multiple threads you can implement the ParallelProcessor trait on your arbitrary struct.
For an example of a single-end parallel processor see the parallel example.
use std::fs::File;
use paraseq::{
fastq,
fastx::Record,
parallel::{ParallelProcessor, ParallelReader, ProcessError},
};
#[derive(Clone, Default)]
struct MyProcessor {
// Your processing state here
}
impl ParallelProcessor for MyProcessor {
fn process_record<R: Record>(&mut self, record: R) -> Result<(), ProcessError> {
// Process record in parallel
Ok(())
}
}
fn main() -> Result<(), ProcessError> {
let file = File::open("./data/sample.fastq")?;
let reader = fastq::Reader::new(file);
let processor = MyProcessor::default();
let num_threads = 8;
reader.process_parallel(processor, num_threads)?;
Ok(())
}
Paired-End Processing
Paired end processing is as simple as single-end processing, but involves implementing the PairedParallelProcessor trait on your struct.
For an example of paired parallel processing see the paired example.
use std::fs::File;
use paraseq::{
fastq,
fastx::Record,
parallel::{PairedParallelProcessor, PairedParallelReader, ProcessError},
};
#[derive(Clone, Default)]
struct MyPairedProcessor {
// Your processing state here
}
impl PairedParallelProcessor for MyPairedProcessor {
fn process_record_pair<R: Record>(&mut self, r1: R, r2: R) -> Result<(), ProcessError> {
// Process paired records in parallel
Ok(())
}
}
fn main() -> Result<(), ProcessError> {
let file1 = File::open("./data/r1.fastq")?;
let file2 = File::open("./data/r2.fastq")?;
let reader1 = fastq::Reader::new(file1);
let reader2 = fastq::Reader::new(file2);
let processor = MyPairedProcessor::default();
let num_threads = 8;
reader1.process_parallel_paired(reader2, processor, num_threads)?;
Ok(())
}
Interleaved Processing
Interleaved processing is also supported by implementing the InterleavedParallelProcessor trait on your struct.
For an example of interleaved parallel processing see the interleaved example.
use std::fs::File;
use paraseq::{
fastq,
fastx::Record,
parallel::{InterleavedParallelProcessor, InterleavedParallelReader, ProcessError},
};
#[derive(Clone, Default)]
struct MyInterleavedProcessor {
// Your processing state here
}
impl InterleavedParallelProcessor for MyInterleavedProcessor {
fn process_interleaved_pair<R: Record>(&mut self, r1: R, r2: R) -> Result<(), ProcessError> {
// Process interleaved paired records in parallel
Ok(())
}
}
fn main() -> Result<(), ProcessError> {
let file = File::open("./data/interleaved.fastq")?;
let reader = fastq::Reader::new(file);
let processor = MyInterleavedProcessor::default();
let num_threads = 8;
reader.process_parallel_interleaved(processor, num_threads)?;
Ok(())
}
Limitations
-
In cases where records have high variance in size, the buffer size predictions will become inaccurate and the shared
Readeroverflow buffer will incur more copy operations. This may lead to suboptimal performance. -
This library currently does not support multiline FASTA.
-
Each
RecordSetmaintains its own buffer, so memory usage practically scales with the number of threads and record capacity. This shouldn't be a concern unless you're running this on a system with very limited memory.
License
MIT
Contributing
Contributions are welcome! Please feel free to submit a Pull Request.
Similar Projects
This work is inspired by the following projects:
This project aims to be directed more specifically at ergonomically processing of paired records in parallel and is optimized mainly for FASTQ files.
It can be faster than seq_io for some use cases, but it is not as feature-rich or rigorously tested, and it does not support multi-line FASTA files.
If the libraries assumptions do not fit your use case, you may want to consider using seq_io or fastq instead.
Dependencies
~1.2–7MB
~40K SLoC