2 unstable releases
| 0.2.0 | Dec 29, 2023 |
|---|---|
| 0.1.0 | Nov 20, 2023 |
#238 in Biology
160KB
408 lines
to-trans
A high-performance exon/CDS spliced transcriptome builder from fasta + GTF/GFF. This is a command-line tool written in Rust designed to build a transcriptome by using a genome (.fa) and a gene model (.gtf/.gff).
Usage
High-performance transcriptome builder from fasta + GTF/GFF
Usage: to-trans --fasta <FASTA> --gtf <GTF> [OPTIONS]
--Arguments:
-f, --fasta <FASTA> Path to your .fa file
-g, --gtf <GTF/GFF> Path to your .gtf/.gff file
Options:
-m, --mode <MODE> Feature to extract from GTF/GFF file (exon or CDS) [default: exon]
-o, --out <OUT> Path to output file [default: transcriptome.fa].
-t, --threads <THREADS> Number of threads [default: max ncpus]
-h, --help Print help
-V, --version Print version
crate: https://crates.io/crates/to-trans
What's new on v.0.2.0
- Now to-trans is ~2-3s faster!
- A parallel approach is now the main algorithm to assemble transcript sequences
Work coming...
to-trans is intended to grow with time, expanding its options and capabilities. In the next release features like: intron extraction, length-based transcriptomes, chromosome-specific builds, among others are coming!
Install/Build
to install to-trans, do:
- get rust:
curl https://sh.rustup.rs -sSf | shon unix, or go here for other options - run
cargo install to-trans(make sure~/.cargo/binis in your$PATHbefore running it)
to build to-trans, do:
- get rust (as described above)
- run
git clone https://github.com/alejandrogzi/to-trans.git && cd to-trans - run
cargo run --release <FASTA> <GTF/GFF> <MODEL> <OUTPUT>
by default to-trans uses exon mode and sends the output to ./transcriptome.fa
Benchmark
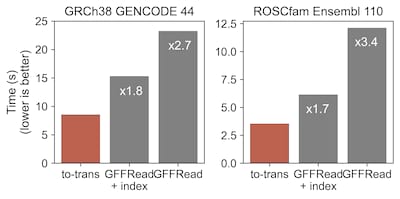
Note that this benchamark is outdated. Now to-trans is ~2-3s faster! For the human genome/gtf, to-trans takes 6 seconds to build a complete transcriptome, that is approximately x3 times faster than GFFRead!
Besides some particular species, such as human (GRCh38) or mouse (GRCm39) that have transcriptomes available, most of the animal kingdom does not count with a pre-defined file with transcript sequences. This becomes a problem when working at the transcript/isoform level.
Compared to GFFRead (1), a gff/gtf utility with a vast range of capabilities, to-trans is able to build a complete transcriptome x2 times faster without the need of indexing the input genome. On the human model, to-trans reaches 8s tops while GFFRead max at 15s with an index (.fai) already available. For the dog, a species without transcript sequences offered in public databases, to-trans takes 3.5s compared to GFFRead doubled times (6s and 12s for indexed and not indexed genomes, respectively).
to-trans offers a novel option to build a transcriptome from a genome + gene model in a very efficent manner. This tool provides high-performance and efficiency without the need of envirnoments or intrincate dependencies, and could be easily attached to workflows/pipelines.
References
- Pertea G and Pertea M. GFF Utilities: GffRead and GffCompare [version 1; peer review: 3 approved]. F1000Research 2020, 9:304 (https://doi.org/10.12688/f1000research.23297.1) https://github.com/gpertea/gffread
Dependencies
~3–11MB
~114K SLoC