2 stable releases
| 1.1.0 | Oct 8, 2021 |
|---|---|
| 1.0.1 | Aug 25, 2021 |
#257 in Biology
Used in gsearch
2MB
2.5K
SLoC
FragGeneScanRs
Installation
From release
Download the build of the latest release for your platform and extract it somewhere in your path.
From source
FragGeneScanRs is written in Rust, so first head over to their
installation instructions. Afterwards, you can install the crate
from crates.io with cargo install frag_gene_scan_rs, or you
can install from here. Clone this repository or download the source code
of the latest release. In this directory, run cargo install --path . to install. The installation progress may prompt you to add a
directory to your path so you can easily execute it.
Usage
You can use FragGeneScanRs with the short options of FragGeneScan but it also provides long-form options and some additional options. It reads from standard input and writes to and standard output by default, allowing shorter calls in case you only need the predicted proteins.
# get predictions for 454 pyrosequencing reads with about 1% error rate
FragGeneScanRs -t 454_10 < example/NC_000913-454.fna > example/NC_000913-454.faa
# get predictions for complete reads
FragGeneScanRs -t complete -w 1 < example/NC_000913.fna > example/NC_000913.faa
Backwards compatible mode
FragGeneScanRs -s seq_file_name -o output_file_name -w [0 or 1] -t train_file_name -p num_threads
where:
-
seq_file_nameis the absolute path for the FASTA file containing DNA sequences that need to undergo gene prediction -
output_file_nameis the absolute path and prefix for the three output files. Files with extensions.out,.faaand.ffnwill be created, respectively containing the gene prediction metadata, protein translations of predicted genes, and the DNA sequences of predicted genes. -
0 or 1for short sequence reads or complete genomic sequences. -
train_file_nameis used to select the training file for one of the following types:completefor complete genomic sequences or short sequence reads without sequencing errorsanger_5for Sanger sequencing reads with about 0.5% error ratesanger_10for Sanger sequencing reads with about 1% error rate454_5for 454 pyrosequencing reads with about 0.5% error rate454_10for 454 pyrosequencing reads with about 1% error rate454_30for 454 pyrosequencing reads with about 3% error rateillumina_5for Illumina sequencing reads with about 0.5% error rateillumina_10for Illumina sequencing reads with about 1% error rate
The corresponding file should be in the subdirectory
trainof the working directory. Other files can be added and selected here. -
num_threadsis the number of threads to be used. Defaults to 1.
Additional options
-
-m meta_file,-n nucleotide_file,-a aa_fileand-g gff_filecan be used to write output to specific files, instead of having the program create filenames with predetermined extentions. These take precedence over the-ooption. -
Leaving out the
-ooption or using the namestdoutcauses FragGeneScanRs to only write the predicted proteins to standard output. The other files can still be requested with the specific options above. -
Leaving out the
-soptions causes FragGeneScanRs to read sequences from standard input. -
-r train_file_dirallows to explicitly specify the pathname of the directory containing the training files, so you can execute the command anywhere on your system. -
The option
-ucan be used for some additional speed and reduced memory when using multithreading. The output will no longer be in the same order as the input (as in FGS and FGS+).
The complete list of options will be printed when running
FragGeneScanRs --help.
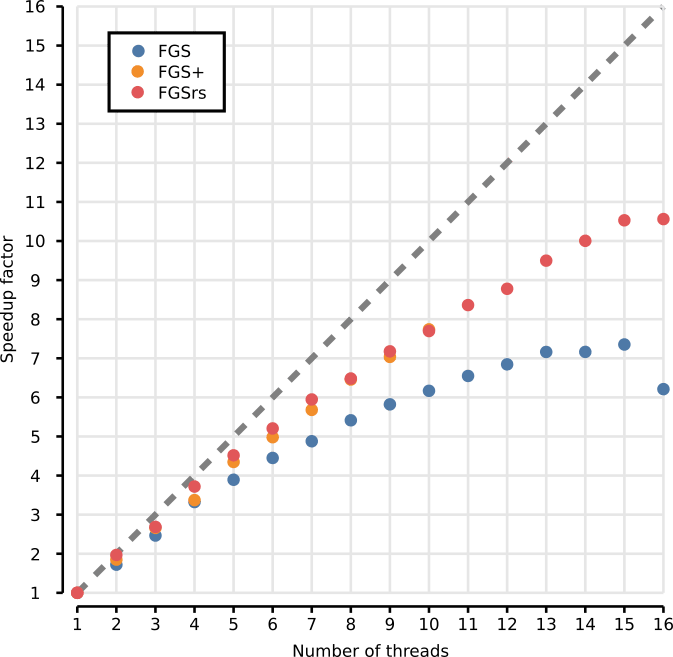
Execution time (version 1.0.0)
Benchmarks were done using the meta/benchmark.sh script on a 16-core
Intel(R) Xeon(R) CPU E5-2650 v2 @ 2.60GHz with 195GB RAM. The datasets
used are the example datasets provided by FragGeneScan. The table
below shows the average execution time of 5 runs. Detailed results
may be found in meta/benchmark.csv. For the short reads (80bp),
FragGeneScanRs is about 22 times faster than FragGeneScan and 1.2
times as fast as FragGeneScanPlus. For the long reads (1328bp) and the
complete genome (Escherichia coli str. K-12 substr. MG1655, 4639675bp),
FragGeneScanRs is 4.3 and 2.2 times faster than FragGeneScan and 1.6 and
233.6 times faster than FGS+.
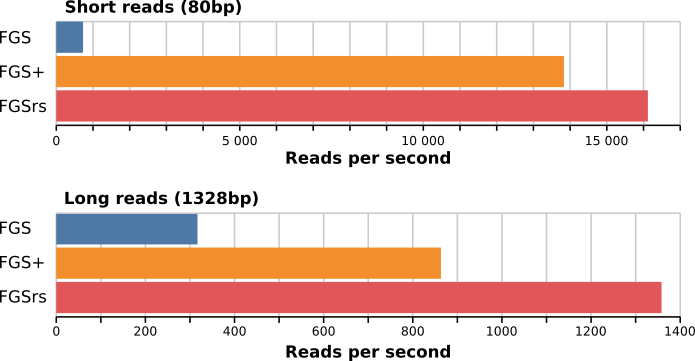
| Short reads | 1 thread | 2 threads | 4 threads | 8 threads | 16 threads |
|---|---|---|---|---|---|
| FragGeneScan | 731 r/s | 1257 r/s | 2158 r/s | 3408 r/s | 3371 r/s |
| FragGeneScanPlus | 13830 r/s | 23997 r/s | 37882 r/s | 54610 r/s | / |
| FragGeneScanRs | 16119 r/s | 29326 r/s | 48593 r/s | 73965 r/s | 99885 r/s |
| Long reads | 1 thread | 2 threads | 4 threads | 8 threads | 16 threads |
|---|---|---|---|---|---|
| FragGeneScan | 317 r/s | 545 r/s | 1053 r/s | 1715 r/s | 1968 r/s |
| FragGeneScanPlus | 863 r/s | 1596 r/s | 2910 r/s | 5573 r/s | / |
| FragGeneScanRs | 1358 r/s | 2674 r/s | 5051 r/s | 8803 r/s | 14343 r/s |
| Complete genome | 1 thread |
|---|---|
| FragGeneScan | 6.668 s |
| FragGeneScanPlus | 712.265 s |
| FragGeneScanRs | 3.049 s |
The commands and arguments used for this benchmarks were:
./FragGeneScan -t 454_10 -s example/NC_000913-454.fna -o example/NC_000913-454 -w 0
./FGS+ -t 454_10 -s example/NC_000913-454.fna -o example/NC_000913-454 -w 0
./FragGeneScanRs -t 454_10 -s example/NC_000913-454.fna -o example/NC_000913-454 -w 0
./FragGeneScan -t complete -s example/contigs.fna -o example/contigs -w 1
./FGS+ -t complete -s example/contigs.fna -o example/contigs -w 1
./FragGeneScanRs -t complete -s example/contigs.fna -o example/contigs -w 1
./FragGeneScan -t complete -s example/NC_000913.fna -o example/NC_000913 -w 1
./FGS+ -t complete -s example/NC_000913.fna -o example/NC_000913 -w 1
./FragGeneScanRs -t complete -s example/NC_000913.fna -o example/NC_000913 -w 1
By default, FragGeneScanPlus outputs only the predicted genes, not the metadata and DNA files. Below are measurements taken when those files aren't generated by FragGeneScanRs either.
| Short reads | 1 thread | 2 threads | 4 threads | 8 threads |
|---|---|---|---|---|
| FragGeneScanPlus | 13765 r/s | 24500 r/s | 39548 r/s | 57147 r/s |
| FragGeneScanRs | 16815 r/s | 28784 r/s | 50157 r/s | 75397 r/s |
The commands used here are:
./FGS+ -t 454_10 -s example/NC_000913-454.fna -o stdout -w 0 > /dev/null
./FragGeneScanRs -t 454_10 -s example/NC_000913-454.fna -o stdout -w 0 > /dev/null
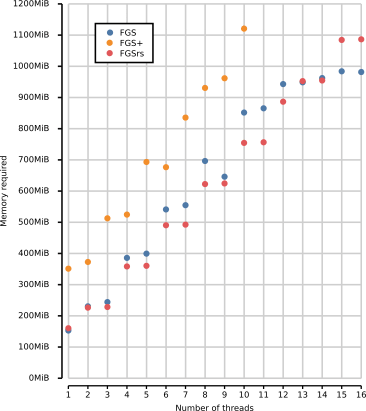
Memory usage (version 1.0.0)
The figure below shows the memory footprint for multithreaded execution
of FGS, FGS+ and FGSrs on long reads (1328 bp). Total memory footprint
(heap, stack and memory-mapped file I/O) is measured using the Massif
heap profiler of Valgrind with the --pages-as-heap option. Race
conditions consistently halt the execution of FGS+ above 10 threads. FGS
and FGSrs generate DNA sequences, protein translations and metadata,
whereas FGS+ only generates protein translations because the software
crashes when other output is generated. FGS and FGS+ report gene
predictions out-of-order, where default in-order reporting was used for
FGSrs.
Dependencies
~6MB
~111K SLoC