15 releases
| 0.1.33 | Jul 2, 2023 |
|---|---|
| 0.1.31 | Nov 8, 2022 |
| 0.1.29 | Mar 9, 2022 |
| 0.1.24 | Dec 22, 2021 |
| 0.1.3 |
|
#806 in Command line utilities
100 downloads per month
7MB
2.5K
SLoC
rustybam
![]()
![]()
![]()
![]()
rustybam is a bioinformatics toolkit written in the rust programing language focused around manipulation of alignment (bam and PAF), annotation (bed), and sequence (fasta and fastq) files.
What can rustybam do?
Here is a commented example that highlights some of the better features of rustybam, and demonstrates how each result can be read directly into another subcommand.
rb trim-paf .test/asm_small.paf `#trims back alignments that align the same query sequence more than once` \
| rb break-paf --max-size 100 `#breaks the alignment into smaller pieces on indels of 100 bases or more` \
| rb orient `#orients each contig so that the majority of bases are forward aligned` \
| rb liftover --bed <(printf "chr22\t12000000\t13000000\n") `#subsets and trims the alignment to 1 Mbp of chr22.` \
| rb filter --paired-len 10000 `#filters for query sequences that have at least 10,000 bases aligned to a target across all alignments.` \
| rb stats --paf `#calculates statistics from the trimmed paf file` \
| less -S
Usage
rustybam [OPTIONS] <SUBCOMMAND>
or
rb [OPTIONS] <SUBCOMMAND>
Subcommands
The full manual of subcommands can be found on the docs.
SUBCOMMANDS:
stats Get percent identity stats from a sam/bam/cram or PAF
bed-length Count the number of bases in a bed file [aliases: bedlen, bl, bedlength]
filter Filter PAF records in various ways
invert Invert the target and query sequences in a PAF along with the CIGAR string
liftover Liftover target sequence coordinates onto query sequence using a PAF
trim-paf Trim paf records that overlap in query sequence [aliases: trim, tp]
orient Orient paf records so that most of the bases are in the forward direction
break-paf Break PAF records with large indels into multiple records (useful for
SafFire) [aliases: breakpaf, bp]
paf-to-sam Convert a PAF file into a SAM file. Warning, all alignments will be marked as
primary! [aliases: paftosam, p2s, paf2sam]
fasta-split Reads in a fasta from stdin and divides into files (can compress by adding
.gz) [aliases: fastasplit, fasplit]
fastq-split Reads in a fastq from stdin and divides into files (can compress by adding
.gz) [aliases: fastqsplit, fqsplit]
get-fasta Mimic bedtools getfasta but allow for bgzip in both bed and fasta inputs
[aliases: getfasta, gf]
nucfreq Get the frequencies of each bp at each position
repeat Report the longest exact repeat length at every position in a fasta
suns Extract the intervals in a genome (fasta) that are made up of SUNs
help Print this message or the help of the given subcommand(s)
Install
conda
mamba install -c bioconda rustybam
cargo
cargo install rustybam
Pre-complied binaries
Download from releases (may be slower than locally complied versions).
Source
git clone https://github.com/mrvollger/rustybam.git
cd rustybam
cargo build --release
and the executables will be built here:
target/release/{rustybam,rb}
Examples
PAF or BAM statistics
For BAM files with extended cigar operations we can calculate statistics about the aliment and report them in BED format.
rustybam stats {input.bam} > {stats.bed}
The same can be done with PAF files as long as they are generated with -c --eqx.
rustybam stats --paf {input.paf} > {stats.bed}
PAF liftovers
I have a
PAFand I want to subset it for just a particular region in the reference.
With rustybam its easy:
rustybam liftover \
--bed <(printf "chr1\t0\t250000000\n") \
input.paf > trimmed.paf
But I also want the alignment statistics for the region.
No problem, rustybam liftover does not just trim the coordinates but also the CIGAR
so it is ready for rustybam stats:
rustybam liftover \
--bed <(printf "chr1\t0\t250000000\n") \
input.paf \
| rustybam stats --paf \
> trimmed.stats.bed
Okay, but Evan asked for an "align slider" so I need to realign in chunks.
No need, just make your bed query to rustybam liftoff a set of sliding windows
and it will do the rest.
rustybam liftover \
--bed <(bedtools makewindows -w 100000 \
<(printf "chr1\t0\t250000000\n") \
) \
input.paf \
| rustybam stats --paf \
> trimmed.stats.bed
You can also use rustybam breakpaf to break up the paf records of indels above a certain size to
get more "miropeats" like intervals.
rustybam breakpaf --max-size 1000 input.paf \
| rustybam liftover \
--bed <(printf "chr1\t0\t250000000\n") \
| ./rustybam stats --paf \
> trimmed.stats.bed
Yeah but how do I visualize the data?
Try out SafFire!
Align once
At the boundaries of CNVs and inversions minimap2 may align the same section of query sequence to multiple stretches of the target sequence. This utility uses the CIGAR (must use --eqx) strings of PAF alignments to determine an optimal split of the alignments such no query base is aligned more than once. To do this the whole PAF file is loaded in memory and then overlaps are removed starting with the largest overlapping interval and iterating.
rb trim-paf {input.paf} > {trimmed.paf}
Here is an example from the NOTCH2NL region comparing CHM1 against CHM13 before trimming:
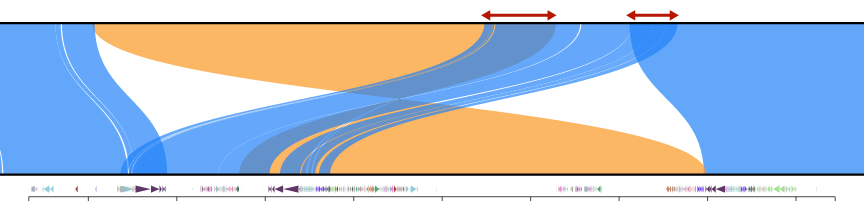
and after trimming
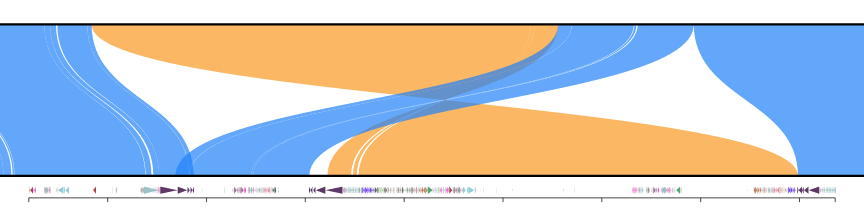
Split fastx files
Split a fasta file between stdout and two other files both compressed and uncompressed.
cat {input.fasta} | rustybam fasta-split two.fa.gz three.fa
Split a fastq file between stdout and two other files both compressed and uncompressed.
cat {input.fastq} | rustybam fastq-split two.fq.gz three.fq
Extract from a fasta
This tools is designed to mimic bedtools getfasta but this tools allows the fasta to be bgzipped.
samtools faidx {seq.fa(.gz)}
rb get-fasta --name --strand --bed {regions.of.interest.bed} --fasta {seq.fa(.gz)}
TODO
- Add a
bedtools getfastalike operation that actually works with bgzipped input.- implement bed12/split
- Allow sam or paf for operations:
- make a sam header from a PAF file
- convert sam record to paf record
- convert paf record to sam record
- make tools seemlessly work with sam and paf
- Add
D4for Nucfreq. - Finish implementing
suns. - Allow multiple input files in
bed-length - Start keeping a changelog
Dependencies
~31–41MB
~639K SLoC