11 releases (7 stable)
| 1.2.1 | Apr 21, 2024 |
|---|---|
| 1.1.0 | Feb 9, 2023 |
| 1.0.3 | Jul 22, 2022 |
| 1.0.1 | Mar 5, 2022 |
| 0.2.1 | Jun 27, 2021 |
#575 in Command line utilities
520KB
12K
SLoC
biodiff
![]()
Compare binary files using alignment algorithms.
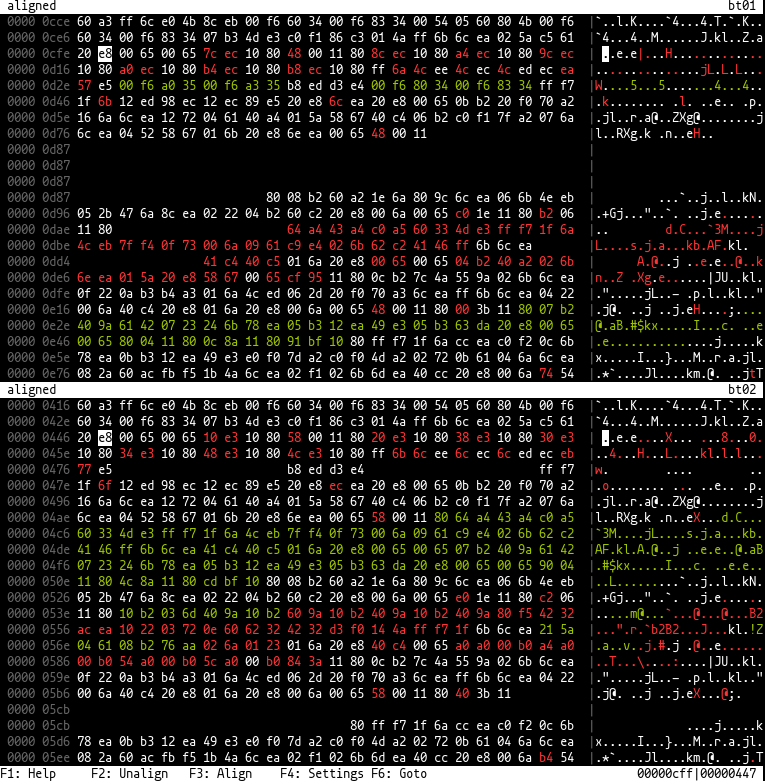
What is this
This is a tool for binary diffing.
The tool is able to show two binary files side by side so that similar places will be at the same position on both sides
and bytes missing from one side are padded.
It uses bio-informatics algorithms from the 'wfa2' or rust-bio library (typically used for DNA sequence alignment) for that.
The dialog boxes for configuration are done using cursive.
Features
- Unaligned view for moving both sides independently as contiguous byte segments
- Aligned view for comparing corresponding bytes of both files
- Many configurable byte representations (bases 2, 8, 10, 16; mixed ascii/hex, braille, roman numerals)
- Right-to-left mode, horizontal and vertical split, ascii and bar column
- Configurable bytes per row, adjustable by pressing
[,],0 - Automatic determination of width by finding repetitions in visible/selected bytes by pressing '='
- Search using text, regex and hexagex
Usage
Execute biodiff file_a file_b in a terminal and you should be dropped into a hex view showing two files side by side.
Initially, the files will not be aligned and displayed without gaps on each side.
By moving the cursor and views to a place where the left side and right side are similar and pressing F3 (or 3), they can be aligned.
This is done block by block in standard configuration, which means that bytes near the cursor are aligned first and further aligned blocks are displayed later on both sides.
It is also possible to do global alignment (of the whole files at once) by changing the settings using F4 (be sure to consult the help on the parameters).
Generally, since it takes quadratic time and space, the global alignment will not work well for files bigger than 64kB.
There is also a "banded" algorithm which is faster, but slightly less accurate.
You can also select a region on one file and by pressing F3 the aligning algorithm will do a semiglobal alignment using the selected bytes as a pattern to find the corresponding bytes on the other file.
It is also possible to print the diff directly to the terminal by using biodiff --print file_a file_b.
In that case (if the files are small enough for it not to take too long), you can add the -gglobal flag to do a global alignment (as opposed to a blockwise one, which is better suited for interactive use).
Installation
If you're lucky, there will be a package available in your primary package manager, see the repology page.
There should be downloadable binary files for some environments under the releases page.
Alternatively, you can also install this using cargo by doing cargo install biodiff.
You will need cmake installed for the wfa2 feature to compile.
Note that in case you use Windows, you need to use the x86_64-unknown-linux-gnu target if you want to have wfa2 support.
You can also execute directly using code from this repository by executing cargo run --release -- file_a file_b.
Note that configuration files are only guaranteed to stay compatible between tagged releases.
By default, settings are stored in a platform-specific user directory.
To use a custom settings directory, set the BIODIFF_CONFIG_DIR environment variable to the desired directory path before running biodiff.
If the directory doesn't exist, it will be automatically created.
License
This project is licensed under the MIT license.
Dependencies
~25–40MB
~547K SLoC