3 releases (breaking)
| 0.3.0 | Jun 20, 2024 |
|---|---|
| 0.2.0 | Jun 13, 2022 |
| 0.1.0 | Nov 4, 2021 |
#232 in Biology
128 downloads per month
105KB
628 lines
![]()
![]()
![]()
![]()
Compute a pairwise SNP distance matrix from one or two alignment(s)
Table of Contents
Motivation
A key point of difference for psdm is the pairwise SNP distance between two
alignment files. This is particularly beneficial if you have computed a SNP distance
matrix for many samples already and want to update the distances with some new samples -
without rerunning the analysis for all samples in the original file.
Another potential use is having two alignment files for the same samples but with sequences generated by different techniques. For example, if you have produced consensus sequences from SNP calls from Illumina and Nanopore and want to see how similar the Illumina-to-Nanopore (inter-technology) distances are - compared to the intra-technology distances.
Despite these motivations, psdm can still be used to compute a "traditional" pairwise
SNP distance matrix for a single FASTA alignment file.
Install
cargo
![]()
Prerequisite: rust toolchain (min. v1.55.0)
$ cargo install psdm
conda
![]()
Prerequisite: conda (and bioconda channel correctly set up)
$ conda install psdm
Precompiled binaries
tl;dr: Run the following snippet to download the latest binary for your system to the current directory and show the help menu.
version="0.3.0"
OS=$(uname -s)
if [ "$OS" = "Linux" ]; then
triple="x86_64-unknown-linux-musl"
elif [ "$OS" = "Darwin" ]; then
triple="x86_64-apple-darwin"
else
echo "ERROR: $OS not a recognised operating system"
fi
if [ -n "$triple" ]; then
URL="https://github.com/mbhall88/psdm/releases/download/${version}/psdm-${version}-${triple}.tar.gz"
wget "$URL" -O - | tar -xzf -
./psdm --help
fi
These binaries do not require that you have the rust toolchain installed.
Currently, there are two pre-compiled binaries available:
- Linux kernel
x86_64-unknown-linux-musl(works on most Linux distributions I tested) - OSX kernel
x86_64-apple-darwin(works for any post-2007 Mac)
An example of downloading one of these binaries using wget
$ version="0.3.0"
$ URL="https://github.com/mbhall88/psdm/releases/download/${version}/psdm-${version}-x86_64-unknown-linux-musl.tar.gz"
$ wget "$URL" -O - | tar -xzf -
$ ./psdm --help
If these binaries do not work on your system please raise an issue and I will potentially add some additional target triples.
homebrew
Prerequisite: homebrew
The homebrew installation is done via the homebrew-bio tap.
$ brew install brewsci/bio/psdm
Container
Docker images are hosted at quay.io.
singularity
Prerequisite: singularity
$ URI="docker://quay.io/mbhall88/psdm"
$ singularity exec "$URI" psdm --help
The above will use the latest version. If you want to specify a version then use a tag (or commit) like so.
$ VERSION="0.3.0"
$ URI="docker://quay.io/mbhall88/psdm:${VERSION}"
docker
Prerequisite: docker
$ docker pull quay.io/mbhall88/psdm
$ docker run quay.io/mbhall88/psdm psdm --help
You can find all the available tags on the quay.io repository.
Local
Prerequisite: rust toolchain
$ git clone https://github.com/mbhall88/psdm.git
$ cd psdm
$ cargo build --release
$ target/release/psdm --help
# if you want to check everything is working ok
$ cargo test
Usage
Quick
Single alignment file
aln1.fa
>s1
ABCDEFGH
>s2
aBN-XFnH
>s0
AbCdEfG-
$ psdm aln1.fa
,s1,s2,s0
s1,0,1,0
s2,1,0,1
s0,0,1,0
Two alignment files
aln2.fa.gz
>s2
xXNNfoo=
>s5 description
AB-DEFGG
$ psdm aln1.fa aln2.fa.gz
,s1,s2,s0
s2,6,5,5
s5,1,2,0
The column names represent the first alignment file provided.
Full
I'd like the sequences to be sorted by identifier in the output
$ psdm -s aln1.fa
,s0,s1,s2
s0,0,0,1
s1,0,0,1
s2,1,1,0
I want a tab-delimited (TSV) matrix instead of a comma-delimited (CSV) one
$ psdm -d "\t" aln1.fa
s1 s2 s0
s1 0 1 0
s2 1 0 1
s0 0 1 0
The case of nucleotides matters to me - i.e., acgt is not the same as ACGT
$ psdm -c aln1.fa
,s1,s2,s0
s1,0,3,3
s2,3,0,5
s0,3,5,0
By default, psdm ignores N's and gaps (-). However, maybe you also want to ignore
X's
$ psdm -e NX- aln1.fa
,s1,s2,s0
s1,0,0,0
s2,0,0,0
s0,0,0,0
Or maybe you don't want to ignore anything
$ psdm -e "" aln1.fa
,s1,s2,s0
s1,0,5,4
s2,5,0,8
s0,4,8,0
I'm impatient, use all the threads I have!
$ psdm -t 0 aln1.fa
Give me long-form output instead of a matrix
$ psdm -l aln1.fa
s1,s1,0
s1,s2,1
s1,s0,0
s2,s1,1
s2,s2,0
s2,s0,1
s0,s1,0
s0,s2,1
s0,s0,0
I'd like to know the progress of the pairwise comparisons
$ psdm -P -t 8 aln.fa
[2024-06-20T02:50:32Z INFO psdm] Using 8 thread(s)
[2024-06-20T02:50:32Z INFO psdm] Loading first alignment file...
[2024-06-20T02:50:38Z INFO psdm] Loaded 200 sequences with length 4411532bp
[2024-06-20T02:50:38Z INFO psdm] Calculating 20100 pairwise distances...
Progress: 50.00% (10050 / 20100)
Write the matrix to a file please
$ psdm -o dists.csv aln1.fa
$ psdm --help
psdm 0.3.0
Michael Hall <michael@mbh.sh>
Compute a pairwise SNP distance matrix from one or two alignment(s)
USAGE:
psdm [OPTIONS] [ALIGNMENTS]...
ARGS:
<ALIGNMENTS>...
FASTA alignment file(s) to compute the pairwise distance for.
Providing two files will compute the distances for all sequences in one file against all
sequences from the other file - i.e., not between sequences in the same file. The first
file will be the column names, while the second is the row names. The alignment file(s)
can be compressed.
OPTIONS:
-c, --case-sensitive
Case matters - i.e., dist(a, A) = 1
-d, --delim <DELIMITER>
Delimiting character for the output table
[default: ,]
-e, --ignored-chars <IGNORED_CHARS>
String of characters to ignore - e.g., `-e N-` -> dist(A, N) = 0 and dist(A, -) = 0
Note, if using `--case-sensitive` the upper- and lower-case form of a character is
needed. To not ignore any characters, use `-e ''` or `-e ""`
[default: N-]
-h, --help
Print help information
-l, --long
Output as long-form ("melted") table
By default the output is a N x N or N x M table where N is the number of sequences in
the first alignment and M is the number of sequences in the (optional) second alignment.
-o, --output <OUTPUT>
Output file name [default: stdout]
-P, --progress
Show a progress bar
-q, --quiet
No logging (except progress info if `-P` is given)
-s, --sort
Sort the alignment(s) by ID
-t, --threads <THREADS>
Number of threads to use. Setting to 0 will use all available
[default: 1]
-V, --version
Print version information
Benchmark
We benchmark against snp-dists (v0.8.2). snp-dists is a brilliant tool
for computing SNP distance matrices and is the inspiration for psdm.
As snp-dists doesn't allow inter-alignment comparisons, we will benchmark the
intra-alignment mode. We use an alignment file of 151 sequences with a length of
4411532bp.
The benchmark times were recorded with hyperfine with 10 runs for each
tool/threads combination.
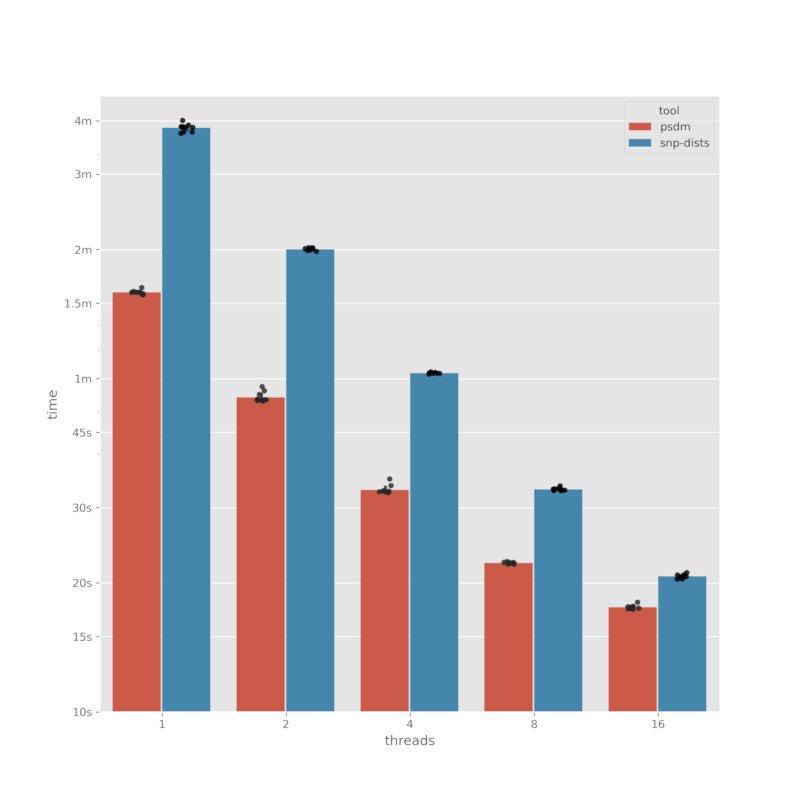
| tool | threads | mean | SD | min | median | max |
|---|---|---|---|---|---|---|
| psdm | 1 | 95.4 | 1.0 | 94.2 | 95.3 | 97.9 |
| snp-dists | 1 | 231.0 | 4.8 | 224.2 | 231.8 | 240.4 |
| psdm | 2 | 54.3 | 1.5 | 53.2 | 53.6 | 57.5 |
| snp-dists | 2 | 120.3 | 0.8 | 118.9 | 120.3 | 121.3 |
| psdm | 4 | 33.0 | 0.8 | 32.5 | 32.7 | 35.0 |
| snp-dists | 4 | 61.8 | 0.2 | 61.5 | 61.8 | 62.2 |
| psdm | 8 | 22.3 | 0.1 | 22.1 | 22.3 | 22.4 |
| snp-dists | 8 | 33.1 | 0.3 | 32.8 | 33.1 | 33.7 |
| psdm | 16 | 17.6 | 0.2 | 17.4 | 17.5 | 18.0 |
| snp-dists | 16 | 20.7 | 0.2 | 20.4 | 20.7 | 21.1 |
Contributing
Contributions are always welcome. For changes to be accepted, they must pass the CI and coverage checks. These include:
- Code is formatted (
cargo fmt). - There are no compiler errors/warnings.
cargo clippy --all-features --all-targets -- -D warnings - Test code coverage has not reduced.
Please also add a succinct description of the contribution in the CHANGELOG.
Cite
psdm is archived at Zenodo.
@software{Hall2021psdm,
author = {Hall, Michael B.},
title = {{psdm: Compute a pairwise SNP distance matrix from
one or two alignment(s)}},
month = nov,
year = 2021,
publisher = {Zenodo},
version = {0.3.0},
doi = {10.5281/zenodo.5706784},
url = {https://doi.org/10.5281/zenodo.5706785}
}
Dependencies
~12–20MB
~281K SLoC