4 releases
| 0.1.3 | Mar 22, 2022 |
|---|---|
| 0.1.2 | Dec 29, 2021 |
| 0.1.1 | Dec 29, 2021 |
| 0.1.0 | Dec 28, 2021 |
#322 in Biology
105KB
1K
SLoC
guide-counter
![]()
![]()
![]()
A better, faster way to count guides in CRISPR screens.
Overview
guide-counter is a tool for processing FASTQ files from CRISPR screen experiments to generate a matrix of per-sample guide counts. It can be used as a faster, more accurate, drop in replacement for mageck count. By default guide-counter will look for guide seqeunces in the reads with 0 or 1 mismatches vs. the expected guides, but can be run in exact matching mode.
Why guide-counter?
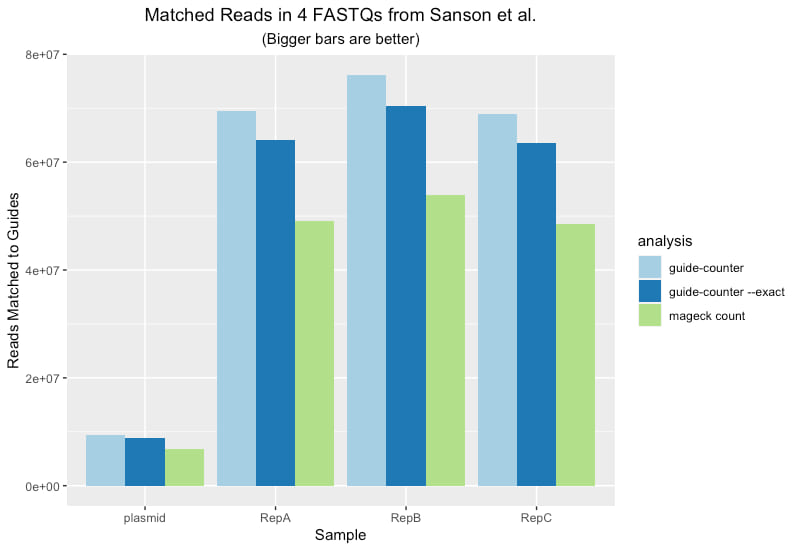
If you have any experience analyzing CRISPR screens you've almost certainly tried mageck. It's widely used, highly cited and generally works well. Surprisingly though, mageck count is both rather slow and misses counting a non-trivial amount of the data.
As an example, we ran data from the Sanson et al paper through both tools. The dataset consists of:
| Sample | Reads | Gzipped FASTQ Size |
|---|---|---|
| Plasmid | 9,821,128 | 377M |
| RepA | 76,471,324 | 2.3G |
| RepB | 85,301,059 | 2.5G |
| RepC | 75,356,900 | 2.2G |
The following plot shows the amount of data recovered per sample by each of three different analyses:
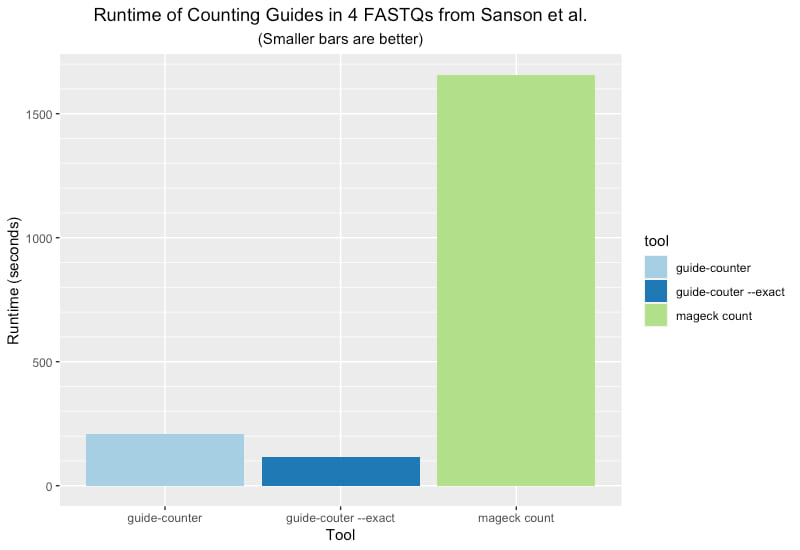
And the following plot shows the runtime for each of the three analyses performed using a single CPU core/thread on an Intel Core i9 powered MacBook Pro laptop:
Installation
Installation can be done using conda:
conda install -c bioconda guide-counter
or with cargo if installed:
cargo install guide-counter
Example Workflow
The following shows an example of running guide-counter followed by mageck test on data from the Sanson et al. 2018 paper:
guide-counter count \
--input plasmid.fq.gz RepA.fq.gz RepB.fq.gz RepC.fq.gz \
--control-pattern control \
--essential-genes metadata/training_essentials.txt \
--nonessential-genes metadata/training_nonessential.txt \
--library metadata/broadgpp-brunello-library-corrected.txt.gz \
--output sanson
mageck test \
--count-table sanson.counts.txt \
--control-id plasmid \
--treatment-id RepA,RepB,RepC \
--norm-method median \
--output-prefix sanson.test
Inputs
The full usage for guide-counter count is reproduced below; this section describes a few of the key inputs in more detail:
| Input Option | Required | Description |
|---|---|---|
--input |
Yes | FASTQ files one per sample. Files may be gzipped or uncompressed. |
--samples |
No | Names for the samples, matched positionally to the FASTQs. If not provided then the input file names minus any `.[fq |
--essential-genes |
No | An optional file of known essential genes. May be gzipped or uncompressed. May be either just gene names, one per line, or tab-delimited with the gene in the first column. If given, guides will be labeled as essential for matching genes, and mean coverage of guides for essential genes computed. |
--nonessential-genes |
No | An optional file of known nonessential genes. May be gzipped or uncompressed. May be either just gene names, one per line, or tab-delimited with the gene in the first column. If given, guides will be labeled as nonessential for matching genes, and mean coverage of guides for nonessential genes computed. |
--control-guides |
No | An optional file of guide IDs for control guides. May be gzipped or uncompressed. May be either just guide IDs, one per line, or tab-delimited data with the guide ID in the first column. If given, matching guides will be labeled as controls, and mean coverage of control guides computed. May be used alone or in conjunction with --control-pattern. |
--control-pattern |
No | An optional regular expression which is applied (case insensitive) to both guide IDs and gene names, and when a match is found, guides are labeled as controls. For example --control-pattern control works well for many human libraries. |
Outputs
The output files are generated:
{output}.counts.txt- a standard count matrix with columns for the guide ID and gene, then one column per sample with raw/unnormalized guide counts.{output}.-extended-counts.txt- an extended version of the counts matrix which includes aguide_typecolumn which will have one of[Essential, Nonessential, Control, Other]per guide as determined based on the gene lists and control information provided.{output}.stats.txt- a file of computed statistics, one row per input sample/FASTQ.
The columns in the stats file are:
| Column | Description |
|---|---|
| file | The path to the input FASTQ file used to generate the stats. |
| label | The label or sample name given to the sample. |
| total_guides | The total number of guides in the guide library (not sample dependent). |
| total_reads | The total number of reads in the input FASTQ file. |
| mapped_reads | The number of reads that could be mapped to a guide. |
| frac_mapped | The fraction of reads (0-1) that could be mapped to a guide. |
| mean_reads_per_guide | The mean number of reads mapped to each guide in the library. |
| mean_reads_essential | The mean number of reads mapped to guides for essential genes. |
| mean_reads_nonessential | The mean number of reads mapped to guides for nonessential genes. |
| mean_reads_control | The mean number of reads mapped to control guides. |
| mean_reads_other | The mean number of reads mapped to other guides (guides not flagged as essential, nonessential or control). |
| zero_read_guides |
Usage
Usage for guide-counter count:
guide-counter-count
Counts the guides observed in a CRISPR screen, starting from one or more FASTQs. FASTQs are one per
sample and currently only single-end FASTQ inputs are supported.
A set of sample IDs may be provided using `--samples id1 id2 ..`. If provided it must have the same
number of values as input FASTQs. If not provided the FASTQ names are used minus any fastq/fq/gz
suffixes.
Automatically determines the range of valid offsets within the sequencing reads where the guide
sequences are located, independently for each FASTQ input. The first `offset-sample-size` reads
from each FASTQ are examined to determine the offsets at which guides are found. When processing the
full FASTQ, checks only those offsets that accounted for at least `offset-min-fraction` of the first
`offset-sample-size` reads.
Matching by default allows for one mismatch (and no indels) between the read sub-sequence and the
expected guide sequences. Exact matching may be enabled by specifying the `--exact-match` option.
Two output files are generated. The first is named `{output}.counts.txt` and contains columns for
the guide id, the gene targeted by the guide and one count column per input FASTQ with raw/un-
normalized counts. The second is named `{output}.stats.txt` and contains basic QC statistics per
input FASTQ on the matching process.
USAGE:
guide-counter count [OPTIONS] --input <INPUT>... --library <LIBRARY> --output <OUTPUT>
OPTIONS:
-c, --control-guides <CONTROL_GUIDES>
Optional path to file with list control guide IDs. IDs should appear one per line and
are case sensitive
-C, --control-pattern <CONTROL_PATTERN>
Optional regular expression pattern used to ID control guides. Pattern is matched, case
insensitive, to guide IDs and Gene names
-e, --essential-genes <ESSENTIAL_GENES>
Optional path to file with list of essential genes. Gene names should appear one per
line and are case sensitive
-f, --offset-min-fraction <OFFSET_MIN_FRACTION>
After sampling the first `offset_sample_size` reads, use offsets that
[default: 0.005]
-h, --help
Print help information
-i, --input <INPUT>...
Input fastq file(s)
-l, --library <LIBRARY>
Path to the guide library metadata. May be a tab- or comma-separated file. Must have a
header line, and the first three fields must be (in order): i) the ID of the guide, ii)
the base sequence of the guide, iii) the gene the guide targets
-n, --nonessential-genes <NONESSENTIAL_GENES>
Optional path to file with list of nonessential genes. Gene names should appear one per
line and are case sensitive
-N, --offset-sample-size <OFFSET_SAMPLE_SIZE>
The number of reads to be examined when determining the offsets at which guides may be
found in the input reads
[default: 100000]
-o, --output <OUTPUT>
Path prefix to use for all output files
-s, --samples <SAMPLES>...
Sample names corresponding to the input fastqs. If provided must be the same length as
input. Otherwise will be inferred from input file names
-x, --exact-match
Perform exact matching only, don't allow mismatches between reads and guides
Dependencies
~10–19MB
~258K SLoC