3 unstable releases
| 0.5.0 | Feb 28, 2023 |
|---|---|
| 0.4.1 | Sep 8, 2022 |
| 0.4.0 | Jan 22, 2021 |
#248 in Biology
255KB
5K
SLoC
Finch
![]()
![]()
Finch is an implementation of min-wise independent permutation locality sensitive hashing ("MinHashing") for genomic data. This repository provides a library and command-line interface that reimplements much of One Codex's existing internal clustering/sequence search tool (and adds new features/extensions!) in Rust.
Getting Started
Installation
You may build Finch from source, which requires Rust >= 1.49. Rust's Cargo package manager (see rustup for Cargo installation instructions) can automatically build and install Finch with cargo install finch_cli.
If you require python bindings, you must take extra steps (see python support). Alternatively, download a prebuilt binary or install from PyPi pip install finch-sketch.
Example Usage
To get started, we first compute sketches for several FASTA or FASTQ files. These sketches are compact, sampled representations of the underlying genomic data, and what allow finch to rapidly estimate distances between datasets. Sketching files uses the finch sketch command:
finch sketch example.fastq example2.fastq
The resulting sketch files (example.fastq.sk and example2.fastq.sk) can then be used with other finch commands (as well as with other MinHash implementations1). Note that all of Finch's commands can take either sketches or raw sequence files. If passed the latter, finch will sketch the files on the fly. Sketches generated on the fly are not saved, however, so you should call finch sketch if you plan to use the sketch multiple times.
Once sketched, multiple sequencing runs can be compared to determine how similar they are:
finch dist example.fastq.sk example2.fastq.sk
This will print results (in JSON) with some key distance statistics, including containment and jaccard similarity scores and a mashDistance distance estimate:
[
{
"commonHashes": 30,
"containment": 0.03,
"jaccard": 0.015228426395939087,
"mashDistance": 0.1669789474914277,
"query": "example2.fastq",
"reference": "example1.fastq",
"totalHashes": 1000
}
]
In this case, these files have an estimated distance of ~0.17 and a containment of 0.03 (i.e., the two FASTQs share 3% of their min-mers). Note that re-computing the sketches with a larger --n-hashes parameter can provide additional resolution for highly similar datasets.
Next, we may want to find the nearest genomes to our example FASTQ across all of RefSeq. To do this, we simply pass our example query file as the first argument and a sketch file containing all the genomes in RefSeq as the second argument (see the Example Data section for pre-computed RefSeq databases that work with finch):
finch dist ./example.fastq.sk ./refseq_sketches_21_1000.sk --max-dist 0.2
Here, we also set a maximum distance of 0.2 in order to filter out less closely related genomes (a distance of 0 would be an identical genome). Setting a maximum ensures that the only relevant results are returned -- omitting this parameter would return distances to all of the genomes in RefSeq.
Note: Each of these commands is detailed further below, and more information is also available by passing the --help flag to each command, e.g., finch dist --help.
Design goals
We have 3 primary design goals with Finch:
-
Support for tracking k-mer/minmer counts;
-
Improved error filtering for use with raw sequence data (i.e., FASTQs); and
-
Improved performance.
Support for counts
Finch is one of the first MinHash implementations to support tracking the abundances of each unique k-mer/minmer.
This allows useful quick interpretations of data (e.g. visualizing sequencing depth with finch hist) in addition to laying the support for more advanced filtering (implemented here) and count-aware distance measures (a future area for investigation and study).
Because Finch also supports the new Mash JSON compatibility format, we're able to save this count information for downstream processing and comparisons.
⚠️ Note that although Finch supports the common JSON interchange format, there may still be incompatibilites due to incompaible hashing seed values (Finch uses a seed of
0, while Mash uses42; see this issue for more discussion).
Filtering
A key design goal for Finch was the ability to accurately – and stably – sketch variable depth sequencing of bacterial isolates (i.e., to not require a high quality assembly). While Mash includes the ability to specify a fixed absolute cutoff (or use a Bloom filter to filter out unique minmers in a sample), in practice this filtering mechanism performs poorly across varied sequencing depths, genome sizes, and sequencing error profiles. The Finch filtering approach implemented here extends several of the ideas in the original Mash paper, as well as prior work on clustering clinical C. diff isolates.
Finch includes two classes of automated filtering (on by default for FASTQs) to address this challenge:
-
Adapative, count-based filtering: Our default implementation "over-sketches" an input file (i.e., includes n fold more minmers than specified by the user) and dynamically determines an appropriate count-based filtering cutoff that removes low abundance k-mers (which predominantly consist of random errors). As a fallback, this implementation never excludes more than an upper bound proportion of minmers that may be reasonably assumed to be erroneous (
ktimes a specified--err-filtererror rate, for a default of 21%). -
Strandedness filtering: We also apply a "strandedness filter" to remove k-mers that are overwhelmingly found in one orientation; in practice these are often seqeuencing adapters or other artifacts that are not representative of the underlying sample (and may erroneously reduce the similarity between two sketches).
In practice, these optimizations dramatically increase the stability and accuracy of distance estimates between variable depth isolates and finished assemblies. The below graph shows the distance between a high quality bacterial assembly and sketches of FASTQ files representing various sequencing depths across 3 filter strategies: no filtering (cutoff=0), "naïve" filtering (cutoff=1), and Finch's adapative, variable-cutoff filtering. We use the containment measure, with results closer to 100% being better:
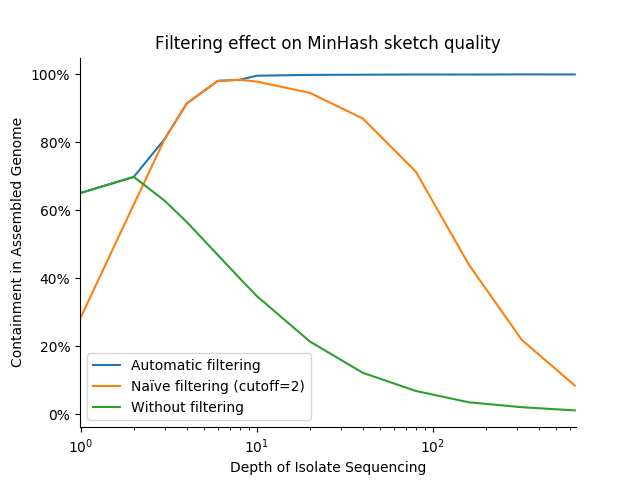
Notably, this strategy performs well across sequencing depths, while fixed cutoffs of 0 and 1 never achieve the near-100% expected containment (and the former fails catastrophically at even modest depths). It is also robust to repeated errors (i.e., error k-mers/minmers with a count >= 2), which can begin to pose a problem at sequencing depths as low a 10-20X.
Speed/performance
In addition to some of the memory safety guarentees that Rust enables, we also see considerable speed gains over existing implementations.
Ideally, hashing should be the rate-limiting step in MinHashing. Profiling indicates finch spends about a third of its time in murmurhash3_x64_128 so we should be within an order of magnitude of this theoretical limit.
| Mash | Mash (filtered) | Sourmash | Finch | Finch (filtered) | |
|---|---|---|---|---|---|
| Time | 238s | 276s | 518s | 99s | 104s |
| Max Memory (Mb) | 1.2 | 501.1 | 13.9 | 21.8 | 60.0 |
Note: Benchmarks run on an Early 2015 Macbook Pro. Benchmark is sketching a 4.8Gb FASTQ file (SRR5132341.fastq) with a sketch size of n=10,000. Finch filtered results use the default options, while Mash uses
-b 500Mbto allocate a 500Mb Bloom filter. Benchmarks used the following commits for Mash (23776db) and sourmash (5da5ee7)
Usage
Finch supports four primary operations, with many of these operations taking similar parameters.
Shared Parameters
Finch can take many parameters to control how sketching is performed (and these options are also available for the dist, hist, and info commands).
-n <N>/--n-hashes <N>controls the overall size of the sketch (higher values give better resolution in comparisons). Default1000.-k <K>/--kmer-length <K>sets the size of the kmers to be hashed (higher values make comparisons much more taxonomically specific). Default21.--seed <S>sets the seed for hashing. This should only be changed if directly exporting sketches for comparison with other versions of the Mash algorithm that use a non-zero default seed. Default0.
After sketching, filtering is performed and can be controlled through several options:
-f/--filter/--no-filterdetermines whether filtering is applied or not (if not specified, filtering is performed for FASTQ files and not performed for FASTA files by default)--min-abun-filter <MIN>/--max-abun-filter <MAX>sets absolute minimum and maximum abundances (inclusive) that all kmers must be present at. The minimum filter will override any adaptive error filter guessing (below).--err-filter <ERR_VALUE>is the default adaptive filtering scheme. ConceptuallyERR_VALUEshould be approximately the error rate of the sequencer or higher. Note that high error rate sequencing data (i.e. long read sequencing) does poorly with MinHashing in general.--strand-filter <V>sets the strand filter cutoff.
Note that if there aren't enough kmers left from the oversketch to satisfy the sketch size, sketching will fail. There are two options that may help:
--oversketch <N>can be used to increase the size of the oversketch (normally 200x) and increase the likelihood that the filtered version will be big enough.--no-strictwill allow Finch to proceed with a sketch smaller than the specified size. Use with caution.
finch sketch
finch sketch will read through a FASTA or FASTQ file and generate a "sketch" that can be used for further .
If the file being read is a sketch and the filtering flag is set (-f/--filter), the abundance filters will be re-applied to the sketch to allow post-sketch filtering.
The strand filters can only be applied to raw FAST(A/Q) files though, as the strand-level data is lost when sketches are saved.
By default sketch generates .sk suffixed files next to the FAST(A/Q) files it sketches.
This can be overridden by passing either -O (capital O) to write to standard out or -o <file> (lowercase o) to write to a file.
To read from standard input, use a filename of -; this allows streaming of files into finch, e.g. cat testfile.fq | finch sketch -o testfile.sk -.
Sketches should be compatible with the original Mash implementation if you edit their src/mash/hash.h and set the hash value to 0 or if you manually override Finch's seed value by setting --seed 42.
finch dist
finch dist will calculate Jaccard distances between different sketches.
If the files listed are FASTA/Q instead of sketches, Finch will automatically sketch them into memory using the same command-line parameters as in sketch or, for files after the first, the parameters used to sketch the first file.
Distances and containments will only be computed from one or more queries to a collection of references; by default the first sketch in the list will be used as the query and all other sketches will be used as references.
This behavior can be manually overriden and other sketches can be used as references by passing --queries <sketch_1>,<sketch_2.
Additionally, passing the --pairwise option will calculate the distances between all sketches and each other.
Due to different counting algoritms and stopping criteria, distances may be slightly different from the calculation in the original Mash program and older version of finch.
Passing the -old-dist flag will revert to the older version of Finch's calculation; support for Mash's exact distance calculation has been dropped as of version 0.3.
finch hist
finch hist will output a histogram in JSON format for each sketch provided.
The histogram is a list of the number of minmers at each depth, e.g. {"sketch_name": [1, 0, 1]} for a sketch with two minmers, one with a depth of 1 (first position) and one with a depth of 3 (third position).
⚠️ You can use the following command with Matplotlib to get a quick histogram:
finch hist test.fastq.sk | python -c 'import json; import matplotlib.pyplot as plt; import sys; v = json.loads(sys.stdin.read()).values()[0]; plt.plot(range(1, len(v)+1), v); plt.show()'. Note that thefinch histJSON output is likely to change in a future version to, e.g., a more compact{count: value}format or similar.
finch info
finch info will output a formatted list of % GC, coverage, etc for each sketch provided.
⚠️ Note that the values returned from this are approximate and the algoritms used to calculate are still rough and liable to change.
Example Data
We've sketched the NCBI RefSeq collection (as of March 27, 2017 using this script) and made tarballs with individual sketches for each bacterial and viral genome available. Links: k=21 and n=1,000, k=31 and n=1,000, k=21 and n=10,000, and k=31 and n=10,000.
References & Notes
There are several other implementations of the Mash algorithm which should be compatible/comparable with this one, notably:
- Mash - First implementation and theoretical paper
- SourMash - Newer implementation in Python; provides a number of experimental features
Notes:
- 1 Please see, however, this issue tracking interoperability and note that other implementations may use a different seed value.
Python Support
You can install Finch using Pip:
pip install finch-sketch
You can compile Finch into a python library with:
pip install maturin
cd lib
maturin build --features=python --release --strip
# or maturin develop, etc
# to cross-compile linux wheels:
cp ../README.md .
# and edit the `readme` key of Cargo.toml
docker run --rm -v $(pwd):/io ghcr.io/pyo3/maturin:main build --features=python --release --strip --interpreter=python3.10 --compatibility=manylinux2010
Then, e.g. to calculate the similarities between two E. coli:
from finch import sketch_file
sketch_one = sketch_file('WIS_EcoB_v2.fas')
sketch_two = sketch_file('WIS_Eco10798_DRAFTv1.fas')
cont, jacc = sketch_one.compare(sketch_two)
Cap'n Proto
There is a finch.capnp in src/serialization file and the output of the MinHash schema (https://github.com/marbl/Mash/blob/54e6d66b7720035a2605a02892cad027ef3231ef/src/mash/capnp/MinHash.capnp)
- the changes by @bovee in https://github.com/bovee/Mash/blob/master/src/mash/capnp/MinHash.capnp
Both are generated after installing capnp and cargo install capnpc with the following command:
capnp compile -orust:finch-lib/src/serialization/ --src-prefix=finch-lib/src/serialization/ finch-lib/src/serialization/finch.capnp
capnp compile -orust:finch-lib/src/serialization/ --src-prefix=finch-lib/src/serialization/ finch-lib/src/serialization/mash.capnp
and then search and replace for the crate:: path to fix it crate::serialization::.
Contributions
Problems or suggestions for improvement can be reported through GitHub issues.
We are happy to accept and/or mentor any additions or fixes (which are best submitted as pull requests).
For our code of conduct, please see CODE_OF_CONDUCT.md.
Dependencies
~7MB
~126K SLoC