38 releases
| 0.8.7 | Apr 6, 2025 |
|---|---|
| 0.8.4 | Dec 30, 2024 |
| 0.8.0 | Nov 30, 2024 |
| 0.7.7 | Jul 19, 2024 |
| 0.4.1 | Sep 9, 2019 |
#52 in Biology
873 downloads per month
Used in nwr
4MB
6.5K
SLoC
intspan
![]()
![]()
![]()
![]()
Install
Current release: 0.8.7
cargo install intspan
cargo install --path . --force #--offline
# or
brew install intspan
# test
cargo test -- --test-threads=1
# local docs
cargo doc --open
# build under WSL 2
mkdir -p /tmp/cargo
export CARGO_TARGET_DIR=/tmp/cargo
cargo build
Concepts
IntSpans
An IntSpan represents sets of integers as a number of inclusive ranges, for example 1-10,19,45-48.
The following figure shows the schema of an IntSpan object. Jump lines are above the baseline; loop lines are below it.
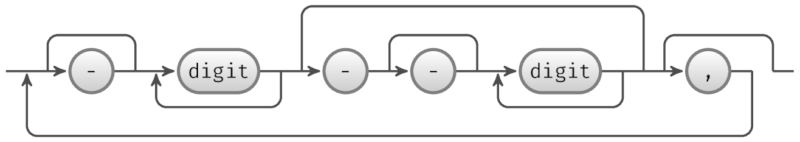
Also, AlignDB::IntSpan and jintspan are implements of the IntSpan objects in Perl and Java, respectively.
Runlists - IntSpans on chromosomes stored in JSON
Very often, we need to deal with many genomic intervals of the same property, e.g., all the exons of a gene, all the promoters of a gene family, all the repeats in a genome, and so on.
Existing formats, such as bedGraph, can partially deal with such situations, but often face
problems of intuitiveness, performance, etc. At the same time, there are only a very limited number
of tools that can handle files in such proprietary formats.
Saving IntSpan to a JSON file is the solution of this toolset, where spanr
handles this job.
- Single:
repeat.json
{
"I": "-",
"II": "327069-327703",
"III": "-",
"IV": "512988-513590,757572-759779,802895-805654,981142-987119,1017673-1018183,1175134-1175738,1307621-1308556,1504223-1504728",
"IX": "-",
"V": "354135-354917",
"VI": "-",
"VII": "778784-779515,878539-879235",
"VIII": "116405-117059,133581-134226",
"X": "366757-367499,712641-713226",
"XI": "162831-163399",
"XII": "64067-65208,91960-92481,451418-455181,455933-457732,460517-464318,465070-466869,489753-490545,817840-818474",
"XIII": "609100-609861",
"XIV": "-",
"XV": "437522-438484",
"XVI": "560481-561065"
}
- Multi:
Atha.json
{
"AT1G01010.1": {
"1": "3631-3913,3996-4276,4486-4605,4706-5095,5174-5326,5439-5899"
},
"AT1G01020.1": {
"1": "5928-6263,6437-7069,7157-7232,7384-7450,7564-7649,7762-7835,7942-7987,8236-8325,8417-8464,8571-8737"
},
"AT1G01020.2": {
"1": "6790-7069,7157-7450,7564-7649,7762-7835,7942-7987,8236-8325,8417-8464,8571-8737"
},
"AT2G01008.1": {
"2": "1025-1272,1458-1510,1873-2810,3706-5513,5782-5945"
},
"AT2G01021.1": {
"2": "6571-6672"
}
}
chr.sizes:S288c.chr.sizes
Ranges
An example is S288c.rg.
The information presented in this format is very similar to formats such as the BED.
I chose this format because of its compactness, readability, and embeddability into other tab-separated files.
I:1-100
I(+):90-150
S288c.I(-):190-200
II:21294-22075
II:23537-24097
The schema of an Range object is shown below.
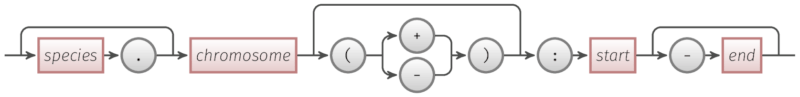
Simple rules:
chromosomeandstartare requiredspecies,strandandendare optional.to separatespeciesandchromosomestrandis one of+and-and surround by round brackets:to separate names and digits-to separatestartandend- For
species:speciesshould be alphanumeric with no spaces, the one exception character is/.- A
speciesis an identity that you can also think of as a strain name, an assembly, or something else.
species.chromosome(strand):start-end
--------^^^^^^^^^^--------^^^^^^----
In this toolset, rgr is used to operate ranges in .rg and .tsv files.
Links of ranges
Types of links:
-
Bilateral links
I(+):13063-17220 I(-):215091-219225 I(+):139501-141431 XII(+):95564-97485 -
Bilateral links with hit strand
I(+):13327-17227 I(+):215084-218967 - I(+):139501-141431 XII(+):95564-97485 + -
Multilateral links
II(+):186984-190356 IX(+):12652-16010 X(+):12635-15993
Synopsis
spanr help
`spanr` operates chromosome IntSpan files
Usage: spanr [COMMAND]
Commands:
genome Convert chr.size to runlists
some Extract some records from a runlist json file
merge Merge runlist json files
split Split a runlist json file
stat Coverage on chromosomes for runlists
statop Coverage on chromosomes for one JSON crossed another
combine Combine multiple sets of runlists in a json file
compare Compare one JSON file against others
span Operate spans in a JSON file
cover Output covers on chromosomes
coverage Output minimum or detailed depth of coverage on chromosomes
gff Convert gff3 to covers on chromosomes
convert Convert runlist file to ranges file
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help
-V, --version Print version
rgr help
`rgr` operates ranges in .rg and .tsv files
Usage: rgr [COMMAND]
Commands:
count Count each range overlapping with other range files
dedup Deduplicate lines in .tsv file(s) based on specified fields or the entire line
field Create/append ranges from fields
filter Filter lines in .tsv files via tests against individual fields
keep Keep the the initial header line(s)
md Convert a .tsv file to a Markdown table
merge Merge overlapped ranges via overlapping graph
pl-2rmp Pipeline - Two Rounds of Merging and Replacing
prop Proportion of the ranges intersecting a runlist file
replace Replace fields in a .tsv file using a replacement map
runlist Filter .rg and .tsv files by comparing with a runlist file
select Select fields in the order listed
sort Sort .rg and .tsv files by a range field
span Operate spans in .tsv/.rg file
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help
-V, --version Print version
File formats
* .rg files are single-column .tsv
* Field numbers in the TSV file start at 1
Subcommand groups:
* Generic .tsv
* dedup / keep / md / replace / filter / select
* Single range field
* field / sort / count / prop / span / runlist
* Multiple range fields
* merge / pl-2rmp
linkr help
`linkr` operates ranges on chromosomes and links of ranges
Usage: linkr [COMMAND]
Commands:
circos Convert links to circos links or highlights
sort Sort links and ranges within links
filter Filter links by numbers of ranges or length differences
clean Replace ranges within links, incorporate hit strands and remove nested links
connect Connect bilateral links into multilateral ones
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help
-V, --version Print version
Examples
spanr
spanr genome tests/spanr/S288c.chr.sizes
spanr genome tests/spanr/S288c.chr.sizes |
spanr stat tests/spanr/S288c.chr.sizes stdin --all
spanr some tests/spanr/Atha.json tests/spanr/Atha.list
spanr merge tests/spanr/I.json tests/spanr/II.json
spanr merge tests/spanr/I.json tests/spanr/II.other.json --all
spanr cover tests/spanr/S288c.rg
spanr cover tests/spanr/dazzname.rg
spanr coverage tests/spanr/S288c.rg -m 2
spanr coverage tests/spanr/S288c.rg -d
spanr gff tests/spanr/NC_007942.gff --tag tRNA
spanr span --op cover tests/spanr/brca2.json
spanr combine tests/spanr/Atha.json
spanr compare \
--op intersect \
tests/spanr/intergenic.json \
tests/spanr/repeat.json
spanr compare \
--op intersect \
tests/spanr/I.II.json \
tests/spanr/I.json \
tests/spanr/II.json
spanr split tests/spanr/I.II.json
spanr stat tests/spanr/S288c.chr.sizes tests/spanr/intergenic.json
spanr stat tests/spanr/S288c.chr.sizes tests/spanr/I.II.json
spanr stat tests/spanr/Atha.chr.sizes tests/spanr/Atha.json
spanr statop \
--op intersect \
tests/spanr/S288c.chr.sizes \
tests/spanr/intergenic.json \
tests/spanr/repeat.json
spanr statop \
--op intersect --all\
tests/spanr/Atha.chr.sizes \
tests/spanr/Atha.json \
tests/spanr/paralog.json
spanr convert tests/spanr/repeat.json tests/spanr/intergenic.json |
spanr cover stdin |
spanr stat tests/spanr/S288c.chr.sizes stdin --all
cargo run --bin spanr convert --longest tests/spanr/repeat.json
spanr merge tests/spanr/repeat.json tests/spanr/intergenic.json |
spanr combine stdin |
spanr stat tests/spanr/S288c.chr.sizes stdin --all
rgr
rgr field tests/Atha/chr.sizes --chr 1 --start 2 -a -s
rgr field tests/spanr/NC_007942.gff -H --chr 1 --start 4 --end 5 --strand 7
rgr field tests/rgr/ctg.tsv --chr 2 --start 3 --end 4 -H -a |
rgr select stdin -H -f length,ID,range > tests/rgr/ctg.range.tsv
rgr sort tests/rgr/S288c.rg
rgr sort tests/rgr/ctg.range.tsv -H -f 3
# ctg:I:1 is treated as a range
rgr sort tests/rgr/S288c.rg tests/rgr/ctg.range.tsv
rgr count tests/rgr/S288c.rg tests/rgr/S288c.rg
rgr count tests/rgr/ctg.range.tsv tests/rgr/S288c.rg -H -f 3
rgr runlist tests/rgr/intergenic.json tests/rgr/S288c.rg --op overlap
rgr runlist tests/rgr/intergenic.json tests/rgr/ctg.range.tsv --op non-overlap -H -f 3
rgr prop tests/rgr/intergenic.json tests/rgr/S288c.rg
rgr prop tests/rgr/intergenic.json tests/rgr/ctg.range.tsv -H -f 3 --prefix --full
rgr merge tests/rgr/II.links.tsv -c 0.95
rgr replace tests/rgr/1_4.ovlp.tsv tests/rgr/1_4.replace.tsv
rgr replace tests/rgr/1_4.ovlp.tsv tests/rgr/1_4.replace.tsv -r
# ctg_2_1_.gc.tsv isn't sorted
cat tests/rgr/ctg_2_1_.gc.tsv | rgr sort stdin | rgr pl-2rmp stdin > /dev/null
cat tests/rgr/II.links.tsv | rgr pl-2rmp stdin
rgr md tests/rgr/ctg.range.tsv --num -c 2
rgr md tests/rgr/ctg.range.tsv --fmt --digits 2
rgr dedup tests/rgr/ctg.tsv tests/rgr/ctg.tsv
rgr dedup tests/rgr/ctg.tsv -f 2
rgr filter tests/spanr/NC_007942.gff -H --str-eq 3:tRNA --str-ne '7:+'
rgr filter tests/spanr/NC_007942.gff -H --case --str-eq 3:trna --str-ne '7:+'
rgr filter tests/rgr/ctg_2_1_.gc.tsv -H --ge 2:0.8
rgr filter tests/rgr/ctg_2_1_.gc.tsv -H --ge 2,2,2:0.8
rgr filter tests/rgr/ctg_2_1_.gc.tsv -H --le 2:0.6 --gt 2:0.45 --eq 3:-1
rgr filter tests/rgr/tn.tsv --ff-eq 1:2
rgr select tests/rgr/ctg.tsv -f 6,1
rgr select tests/rgr/ctg.tsv -H -f ID,1
rgr span tests/rgr/S288c.rg --op trim -n 0
rgr span tests/rgr/S288c.rg --op trim -n 10
rgr span tests/rgr/S288c.rg --op shift --mode 3p -n 10
rgr span tests/rgr/S288c.rg --op flank --mode 3p -n=-1 -a
rgr span tests/rgr/S288c.rg --op excise -f 1 -n 20
rgr span tests/rgr/ctg.range.tsv -H -f 3 -a --op trim -n 100 -m 5p
cat tests/rgr/ctg.range.tsv | sort -k1,1nr
keep-header tests/rgr/ctg.range.tsv tests/rgr/ctg.range.tsv -- sort -k1,1nr
cargo run --bin rgr keep tests/rgr/ctg.range.tsv -- sort -k1,1nr
cargo run --bin rgr keep tests/rgr/ctg.range.tsv tests/rgr/ctg.range.tsv -- wc -l
cat tests/rgr/ctg.range.tsv | cargo run --bin rgr keep tests/rgr/ctg.range.tsv stdin -- wc -l
linkr
linkr sort tests/linkr/II.links.tsv -o tests/linkr/II.sort.tsv
rgr merge tests/linkr/II.links.tsv -v
linkr clean tests/linkr/II.sort.tsv
linkr clean tests/linkr/II.sort.tsv --bundle 500
linkr clean tests/linkr/II.sort.tsv -r tests/linkr/II.merge.tsv
linkr connect tests/linkr/II.clean.tsv -v
linkr filter tests/linkr/II.connect.tsv -n 2
linkr filter tests/linkr/II.connect.tsv -n 3 -r 0.99
linkr circos tests/linkr/II.connect.tsv
linkr circos --highlight tests/linkr/II.connect.tsv
Steps:
sort
|
v
clean -> merge
| /
| /
v
clean
|
V
connect
|
v
filter
S288c
linkr sort tests/S288c/links.lastz.tsv tests/S288c/links.blast.tsv \
-o tests/S288c/sort.tsv
linkr clean tests/S288c/sort.tsv \
-o tests/S288c/sort.clean.tsv
rgr merge tests/S288c/sort.clean.tsv -c 0.95 \
-o tests/S288c/merge.tsv
linkr clean tests/S288c/sort.clean.tsv -r tests/S288c/merge.tsv --bundle 500 \
-o tests/S288c/clean.tsv
linkr connect tests/S288c/clean.tsv -r 0.8 \
-o tests/S288c/connect.tsv
linkr filter tests/S288c/connect.tsv -r 0.8 \
-o tests/S288c/filter.tsv
wc -l tests/S288c/*.tsv
# 229 tests/S288c/clean.tsv
# 148 tests/S288c/connect.tsv
# 148 tests/S288c/filter.tsv
# 566 tests/S288c/links.blast.tsv
# 346 tests/S288c/links.lastz.tsv
# 74 tests/S288c/merge.tsv
# 282 tests/S288c/sort.clean.tsv
# 626 tests/S288c/sort.tsv
cat tests/S288c/filter.tsv |
perl -nla -F"\t" -e 'print for @F' |
spanr cover stdin -o tests/S288c/cover.json
spanr stat tests/S288c/chr.sizes tests/S288c/cover.json -o stdout
Atha
gzip -dcf tests/Atha/links.lastz.tsv.gz tests/Atha/links.blast.tsv.gz |
linkr sort stdin -o tests/Atha/sort.tsv
linkr clean tests/Atha/sort.tsv -o tests/Atha/sort.clean.tsv
rgr merge tests/Atha/sort.clean.tsv -c 0.95 -o tests/Atha/merge.tsv
linkr clean tests/Atha/sort.clean.tsv -r tests/Atha/merge.tsv --bundle 500 -o tests/Atha/clean.tsv
linkr connect tests/Atha/clean.tsv -o tests/Atha/connect.tsv
linkr filter tests/Atha/connect.tsv -r 0.8 -o tests/Atha/filter.tsv
wc -l tests/Atha/*.tsv
# 4500 tests/Atha/clean.tsv
# 3832 tests/Atha/connect.tsv
# 3832 tests/Atha/filter.tsv
# 785 tests/Atha/merge.tsv
# 5416 tests/Atha/sort.clean.tsv
# 7754 tests/Atha/sort.tsv
cat tests/Atha/filter.tsv |
perl -nla -F"\t" -e 'print for @F' |
spanr cover stdin -o tests/Atha/cover.json
spanr stat tests/Atha/chr.sizes tests/Atha/cover.json -o stdout
License

Dependencies
~24–36MB
~546K SLoC