8 releases (5 breaking)
| new 0.7.1 | Apr 29, 2025 |
|---|---|
| 0.7.0 | Apr 6, 2025 |
| 0.6.0 | Mar 23, 2025 |
| 0.5.0 | Feb 15, 2025 |
| 0.2.0 | Dec 11, 2024 |
#37 in Science
234 downloads per month
1MB
25K
SLoC
gorder: Everything you will ever need for lipid order calculations
![]()
Command line tool for calculating lipid order parameters from Gromacs simulations.
Installation
-
Install gorder:
$ cargo install gorder
Usage
- Prepare a yaml file describing the parameters of your analysis. It's as simple as:
structure: system.tpr
trajectory: md.xtc # use your MD trajectory directly - no PBC handling or molecule fixing needed
analysis_type: !AAOrder
heavy_atoms: "element name carbon"
hydrogens: "element name hydrogen"
output: order.yaml
- Run the
gordertool. The program will automatically find bonds between the selected atoms, classify lipid molecules, and calculate order parameters for them.
$ gorder YOUR_INPUT_YAML_FILE
- See the results of the analysis in
order.yaml.
See the gorder manual for more information about using this program.
Features
-
⚛️ Atomistic, coarse-grained, and united-atom systems
Calculates order parameters for individual bonds of individual lipid types in atomistic, coarse-grained, and united-atom systems. -
🔠 Powerful selection language
Allows for simple yet powerful atom selection using a VMD-like selection language, supporting regular expressions and groups from NDX files. -
🔍 Automatic identification of molecule types
Automatically recognizes bonds and classifies molecule types based on their topology. Order parameters are calculated and reported separately for each molecule type. -
📋 Various output formats
Outputs results in YAML, XVG, CSV, and custom "table" format. -
🏗️ Any model and force-field
Is completely force-field agnostic. Martini? CHARMM? Slipids? Berger lipids? Your own toy force-field? As long as your lipids have bonds, it will work. -
🍃 Leaflet-wise analysis
Performs scrambling-safe assignment of lipids to membrane leaflets using four different methods, and then calculates lipid order parameters for individual leaflets. -
🟠 Order parameters for vesicles
Dynamically calculates membrane normals based on the membrane's shape, enabling the calculation of order parameters for highly curved systems such as micelles, tubes, and vesicles. -
🗺️ Order parameter maps
Constructs 2D projections of order parameters, so you know what parts of the membrane are ordered and disordered. -
📉 Error estimation
Automatically estimates the error of the analysis and indicates how well your analysis has converged. -
🌍 Analysis of specific membrane regions
Dynamically selects lipids in a specified part of the membrane and calculates order parameters only for them. -
🔗 Trajectory concatenation
Seamlessly concatenates multiple XTC trajectories during the analysis, in most cases eliminating the need for data preprocessing. -
⚡ Extremely fast
Is extremely fast due to its ability to read only the necessary atoms from XTC files and its support for multithreading. -
👨💻 Python and Rust API
Is also available as a Python package and a Rust crate, allowing seamless integration with Python and/or Rust code. -
📖 Extensive manual
Includes a comprehensive manual detailing how to use the program, along with its strengths and limitations.
Validation
Atomistic order parameters
A CHARMM36m simulation of a membrane consisting of 256 lipids was used to validate the calculation of atomistic order parameters. In total, the system contained ~64,500 atoms. The trajectory was 200 ns long and consisted of 10,000 frames. The following tools were used for validation:
OrderParameter.pyby NMR Lipids- VMD's
calc_op.tclscript order_paramstool provided as a part ofLOOS
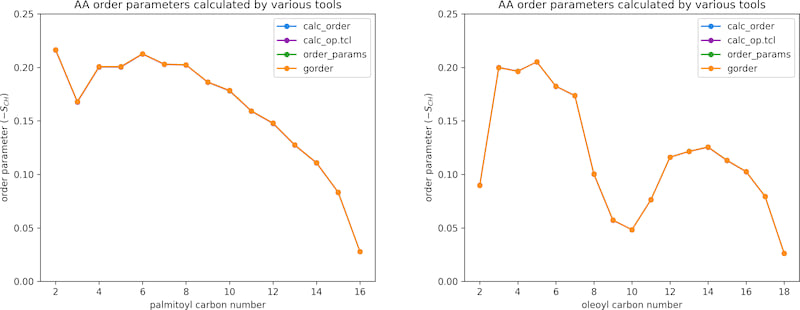
Left chart shows the results for the palmitoyl tail of POPC, the right chart for the oleoyl tail. All tools return the same results.
Coarse-grained order parameters
A Martini 3 simulation of a membrane consisting of 512 POPC lipids was used to validate the calculation of CG order parameters. In total, the system contained ~16,800 beads. The trajectory had a length of 1 μs and consisted of 10,000 frames. The following tools were used for validation:
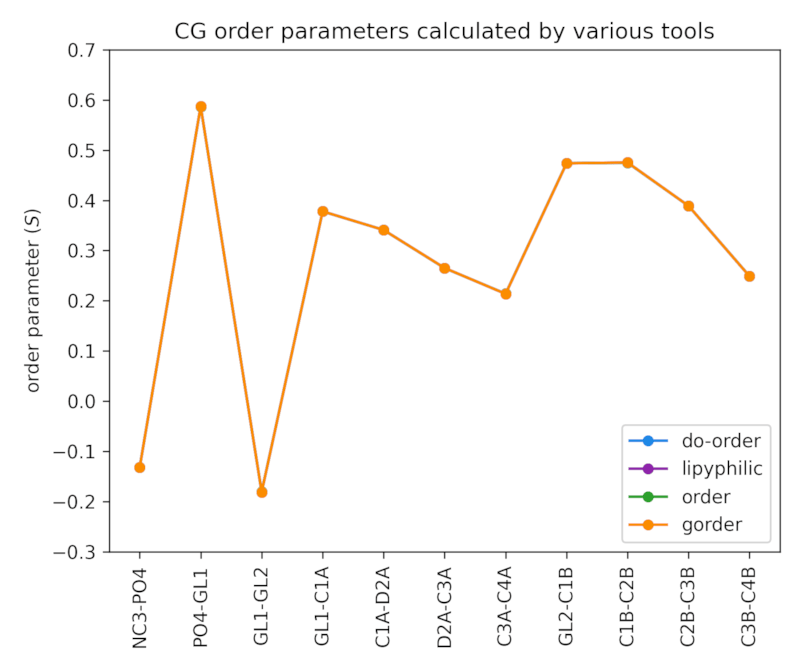
All programs produce the same results. Minor variations due to the employed calculation approaches are too small to be visible in the chart.
United-atom order parameters
A Berger lipids simulation of a membrane obtained from the NMR Lipids DataBank [link to archive] consisting of 256 POPC lipids was used to validate the calculation of UA order parameters. In total, the system contained ~44,300 atoms. The trajectory had a length of 300 ns and consisted of 3,000 frames. The following tools were used for validation:
buildHtoolg_lomeproprogramgmx order(saturated carbons only)
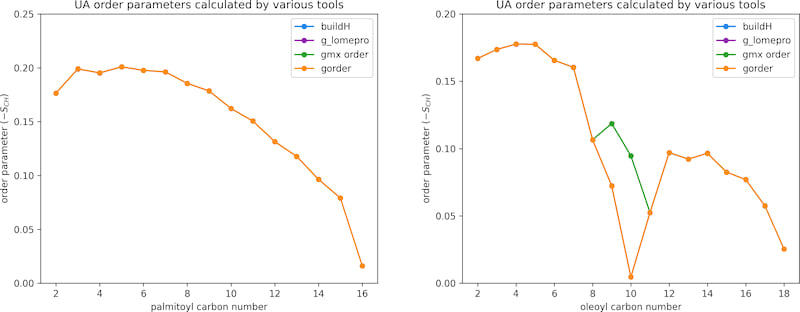
Left chart shows the results for the palmitoyl tail of POPC, the right chart for the oleoyl tail. buildH and gorder generate identical output. g_lomepro also produces the same output, except for the methyl carbon, for which no order parameter is reported. gmx order behaves the same as g_lomepro for the palmitoyl tail, but, as reported previously [link to article], returns incorrect order parameters for unsaturated carbons of the oleoyl tail.
Benchmarking
Atomistic order parameters
Run times of the analyses performed in the Validation section by various tools. We also show the performance of gmx order, even though it actually calculates united-atom order parameters, because it is (unfortunately) commonly used for analyses of all-atom systems as well:
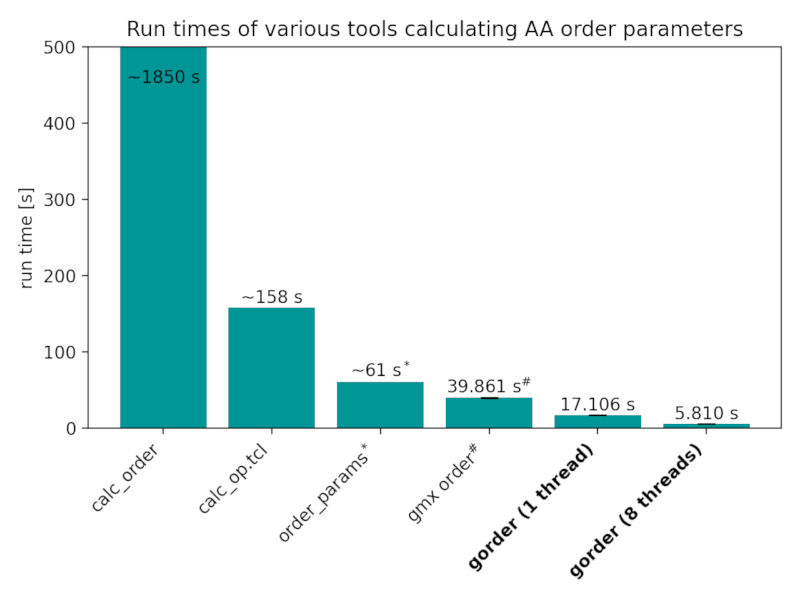
Benchmarks were conducted using gorder version 0.6 on GNU/Linux Mint 20.2 with an 8-core Intel Core i7-11700 CPU and Samsung 870 EVO SSD. The benchmarks were run with a cold cache using hyperfine. Both gorder and gmx order were run 5 times each, while calc_order, calc_op.tcl, and order_params were slower so only 1 run for each was performed.
* Note that order_params requires the analysis to be run twice to obtain full order parameters for both chains. The reported time is for both runs.
# Note that gmx order requires the analysis to be run twice to obtain full order parameters for both chains. More importantly, note that gmx order does not calculate the order parameters correctly. You should not use it for an atomistic system! Even if you only want to calculate lipid order parameters for a single (saturated) chain, gmx order is still much slower than gorder run using a single thread (20.70 seconds vs 11.40 seconds).
Coarse-grained order parameters
Run times of the analyses performed in the Validation section by various tools:
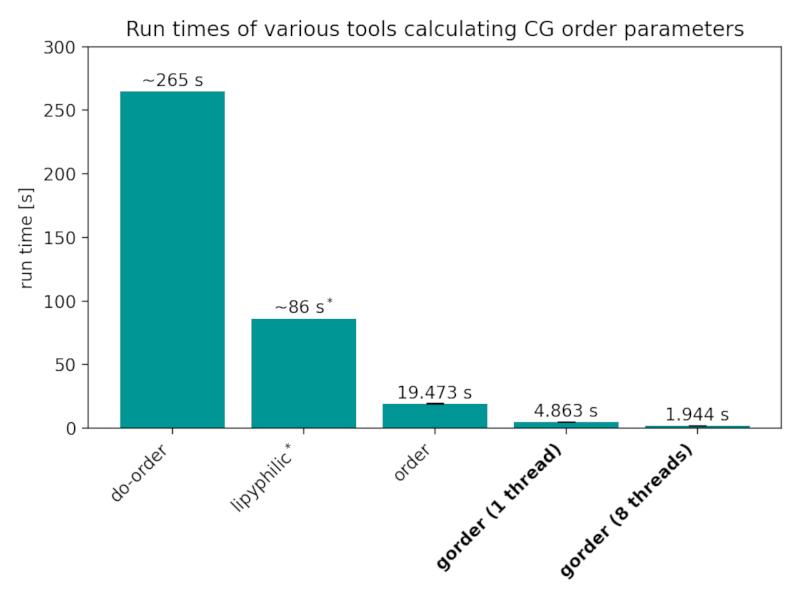
Benchmarks were conducted using gorder version 0.6 on GNU/Linux Mint 20.2 with an 8-core Intel Core i7-11700 CPU and Samsung 870 EVO SSD. The benchmarks were run with a cold cache using hyperfine. Both gorder and order were run 5 times each, while do-order and lipyphilic were dramatically slower so only 1 run for each was performed.
* Note that the lipyphilic library is not able to calculate order parameters for the individual bonds simultaneously, requiring the analysis to be run multiple times. The reported time is the sum of all runs. If you are only interested in average order parameters for one entire lipid tail, lipyphilic is quite fast (though still slower than gorder).
United-atom order parameters
Run times of the analyses perform in the Validation section by various tools:
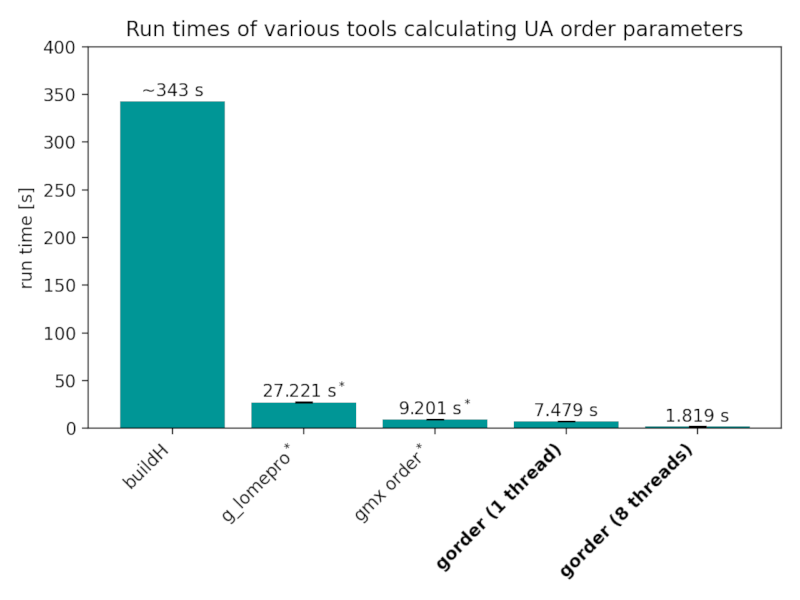
Benchmarks were conducted using gorder version 0.6 on GNU/Linux Mint 20.2 with an 8-core Intel Core i7-11700 CPU and Samsung 870 EVO SSD. The benchmarks were run with a cold cache using hyperfine. gorder, g_lomepro, and gmx order were run 5 times each, while buildH was dramatically slower so only 1 run was performed for it. g_lomepro was run using 8 threads.
* Note that g_lomepro and gmx order require the analysis to be run twice to obtain full order parameters for both chains. The reported times are for both runs. Even if you only want to calculate lipid order parameters for a single (saturated) chain, gmx order is still slightly slower than gorder run using a single thread (4.75 seconds vs 4.16 seconds).
Python and Rust API
gorder is also available as a Python package and a Rust crate. For more information, see the gorder manual, the Python API documentation, or the Rust API documentation.
License
The command line tool, the Python package, and the Rust crate are all released under the MIT License.
Limitations
gorderdoes not fully support simulation boxes that are not orthogonal. You can still use them but it requires some additional effort.gorderdoes not support TPR files generated with Gromacs older than version 5.1. But you can use PDB, GRO, or PQR file instead.gorderis developed for Linux. While it should work on other operating systems, it is not guaranteed.
Dependencies
~24–34MB
~573K SLoC