| 3.0.0 |
|
|---|---|
| 2.0.0 |
|
| 0.3.1 |
|
| 0.2.0 |
|
| 0.1.0 |
|
#5 in #similarity-analysis
745KB
1K
SLoC
BLUTILS
The Blutils tool allow user to run and generate consensus identities of Blast
results. Currently the BlastN is available.
Installation
Blutils package could be installed directly from
crates.io using cargo:
cargo install blutils
After installed, Blutils could be evoked using the blu command.
blu --help
The output should be close to:
A utility to make it easier to run and analyze Blast results
Usage: blu <COMMAND>
Commands:
blast Execute the parallel blast and run consensus algorithm
check Check `Blutils` dependencies
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help information
-V, --version Print version information
Check dependencies
Optionally you could check OS dependencies before run Blutils. Naturally BLutils
depends on Ncbi-Blast+ tool to be installed on the host system to perform
parallel blast search. To check if the host OS has these package installed run
the Blutils checker for linux systems:
blu check linux
Note: Currently the system check is available only for linux systems and assumes that dependencies could be evoked directly from terminal.
Run Blast with Blutils
The Blutils execution is simple. To check all available options evoke the
blast subcommand help:
blu blast run-with-consensus --help
After inspect available options, simple run Blutils with test data. First
download test data from the project github directory:
export INPUT_DIR=https://raw.githubusercontent.com/sgelias/blutils/main/test/mock/input
curl ${INPUT_DIR}/query/query.fna > query.fna
curl ${INPUT_DIR}/query/ref_databases/mock-16S.fna > mock-16S.fna
curl ${INPUT_DIR}/query/ref_databases/mock-16S_taxonomies.tsv > mock-16S_taxonomies.tsv
Then run Blutils:
blu blast run-with-consensus \
query.fna \
mock-16S.fna \
mock-16S_taxonomies.tsv \
output \
-t 6 \
--taxon bacteria \
--strategy relaxed \
-f
Seems the above commands, the output files could be found in output directory
which will contains two additional files named blast.out and
blutils.consensus.json. The first contains default Blast tabular response and
the former, the Blutils response, which will be close to:
[
{
"query": "NR114924.257984.Bac",
"taxon": {
"rank": "class",
"taxid": 1760,
"percIdentity": 100.0
}
},
{
"query": "NR025123.135626.Bac",
"taxon": {
"rank": "species",
"taxid": 135626,
"percIdentity": 100.0
}
},
{
"query": "INVALID_SEQUENCE",
"taxon": null
}
]
Blast execution
Blast execution try to reaches the full available CPU saturation. At the default
multithread blast execution mode, the full saturation is not reached. To run
Blast through Blutils it is possible. All the steps taken during this process
can be seen in the image below.
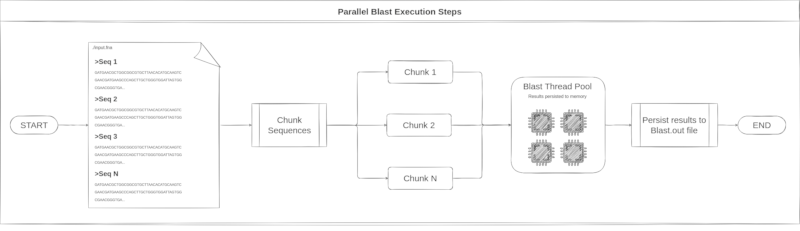
Consensus generation
Different from consensus generations from QIIME
2, the Blutils consensus algorithm performs
a data pre-filtering based on Blast results for bit-score and perc-identity,
seems the algorithm described in the image below.
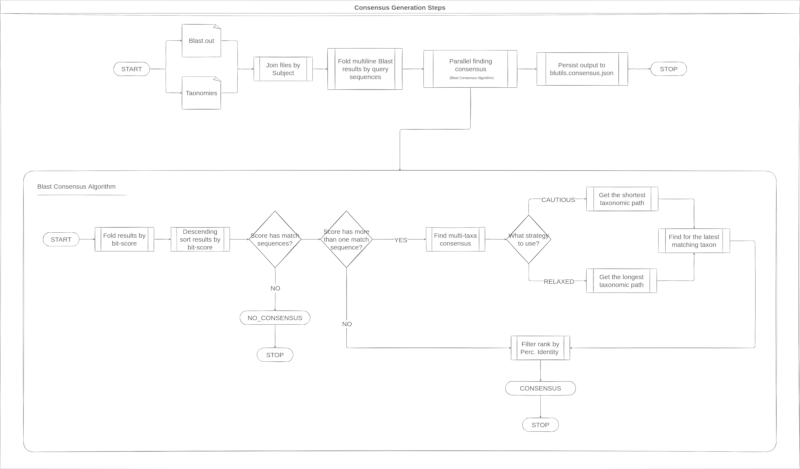
Next steps
This project contains only basic features to run BlastN and generate consensus identities. Thus, many features should be created, like create the database extractor to get data from official NCBI taxdump results and build FASTA database simultaneously, and others. We welcome new feature suggestions when needed!
Dependencies
~25–36MB
~610K SLoC