10 releases
| 0.4.1 | Apr 11, 2025 |
|---|---|
| 0.4.0 | Feb 16, 2024 |
| 0.3.0 | Oct 30, 2023 |
| 0.2.5 | Aug 31, 2023 |
| 0.1.0 | Jun 7, 2023 |
#16 in Biology
142 downloads per month
130KB
2.5K
SLoC
Align-cli
A tool to help your manual mass spec inspection workflow. With alignments, isobaric sequences, and other mass spec information.
Installing
Using winget
On windows use winget install --id Snijderlab.AlignCli.
From binary
- Pick the correct binary for your machine in the release
- Place it in a nice location on your machine
- If you are using a unix-based operating system (linux or mac) do not forget to
chmod +x <binary> - Open a terminal and use the tool
- If you want you can add the location of the binary to your path, this makes it so that you can use it across your whole machine
More elaborate instructions for Windows (installing another program)
With cargo
- First install Rust.
- Install the tool using cargo (part of Rust)
cargo install align-cli
From source
- First install Rust.
- Clone the repository.
- Build with cargo
cargo build --release - Or, if you want to install
cargo install --path ..
Quick usage overview
- Pairwise alignment
- Align two sequences
align <A> <B>, this shows the best alignment for these two sequences. - Align a single peptide to a database
align <A> --file <FILE.fasta>. - Align a single peptide to the IMGT database
align <A> --imgt. - Align a single peptide to the V-J-C domains in the IMGT database
align <A> --domain. - Align a single peptide to a specific gene in IMGT database
align <A> --specific-gene <GENE>. - For any of these you can control if the peptides have to allign fully (
--global), if you want to see the best possible subsequence alignment (--local), or a more elaborate mode (see--helpand--type).
- Align two sequences
- Get information about a single sequence
align <sequence>, this shows many basic properties (like mass) and generates isobaric sequences to this sequence.- Use
--fixed <MODIFICATIONS>and--variable <MODIFICATIONS>to fine tune the generated isobaric sequences.
- Use
- Get information about a single modification
align --modification <MODIFICATION>.- Use a full name to list its properties eg
--modification Oxidation - Use a formula to find all modifications with that formula eg
--modification Formula:O - Use a mass to find all modifications with that mass eg
--modification +15.995
- Use a full name to list its properties eg
- List IMGT genes
align --imgtoralign --specific-gene <GENE>.
For all additional options and more description use align --help.
Example usage


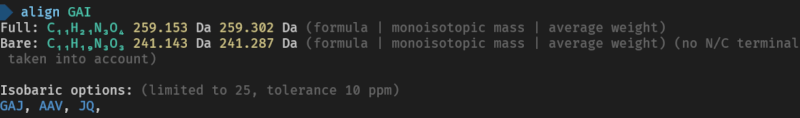
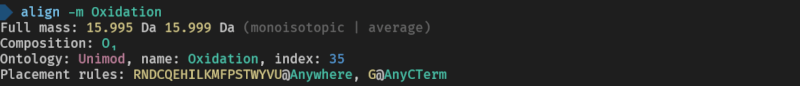

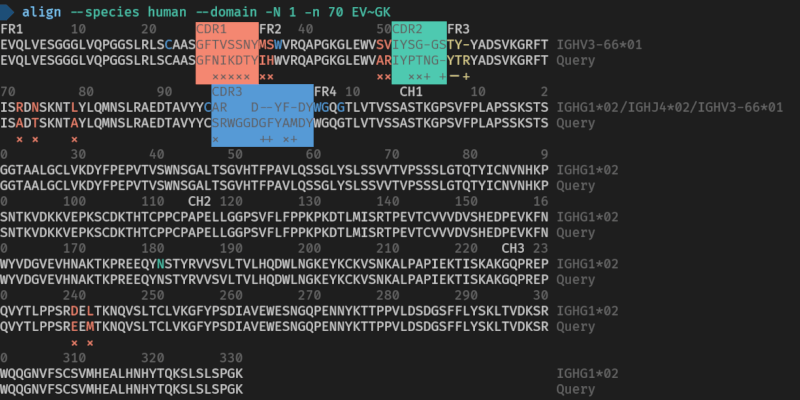
Here are the used commands for reference
> align AKTNLSHLGYGMDV AKEGGLHSIGYGMDV
> align AKTNLNHLGY BHJGYGMDV --local --context
> align GAI
> align --modification 23 --tolerance 0.5da
> align -m Oxidation
> align --species human --domain -N 1 -n 70 EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
Dependencies
~20–28MB
~338K SLoC